








Coronary artery spasm (CAS) is an established cause for anginal chest pain, the cardinal symptom of myocardial ischaemia, in patients with angiographically unobstructed coronary arteries. Evidence from large clinical studies has revealed that about 50% of patients undergoing diagnostic coronary angiography for suspected coronary artery disease (CAD) had either normal or near normal coronary arteries (<20% stenosis) or non-obstructive CAD (<50% stenosis).1,2 Abnormal coronary vasomotion occurred in about 60% of patients with unobstructed coronary arteries who underwent acetylcholine (ACh) provocation testing during coronary angiography, an established method for the assessment of CAS.3,4 These functional vasoconstrictor disorders may occur at the epicardial level, known as focal or diffuse pronounced epicardial spasm, and/or in the microvasculature, known as microvascular spasm.2
An overview of the current diagnostic criteria of CAS is shown in Table 1.5,6 Due to the heterogeneous clinical presentation of these patients regarding rest- and/or exercise-related angina, severity and frequency of symptoms, presence of coronary atherosclerosis, spasm localisation, the diagnosis and treatment of CAS are still challenging. The lack of a consistent definition of epicardial spasm in previous studies, for example regarding the degree of vasoconstriction for a positive CAS provocation test, further impedes the understanding of the underlying pathophysiological mechanisms. Nevertheless, several mechanisms have been proposed based on experimental and clinical studies. Central players involved in all scenarios are endothelial cells and vascular smooth muscle cells (VSMC), which can be influenced by extrinsic factors such as oxidative stress and inflammation (often induced by established cardiovascular risk factors, such as hypertension, hypercholesterinaemia, diabetes, obesity and smoking), as well as genetic mutations/polymorphisms and the autonomic nervous system (Figure 1).7–9 The interplay of both the endothelium and the VSMC layer plays a crucial role in the regulation of coronary vascular tone and two schools of thought exist that see either endothelial dysfunction or VSMC hyperreactivity as primary contributors to CAS.8,10,11 The aim of this article is to summarise the current knowledge and possible interplay of an impaired endothelial and VSMC function in the pathogenesis of CAS.
Normal and Abnormal Coronary Vasomotor Function
Under physiological conditions, the interplay between the endothelium and the VSMC layer is crucial for adequate regulation of coronary vascular tone. Both cell types are influenced by various stimuli derived from haemodynamic (e.g. blood pressure and blood flow) and metabolic (e.g. adenosine and oxygen partial pressure) changes, as well as by the autonomic innervation and circulating vasoactive substances ,such as ACh, adenosine, serotonin and histamine, acting differently on the endothelium and VSMCs. The endothelium is much more than a passive barrier between blood and the vessel wall. By acting as signal transducers of haemodynamic forces (caused by the blood flow) and circulating substances, endothelial cells contribute to coronary tone modulation. In response to these vasoactive stimuli, endothelial cells are capable of synthesising and releasing a variety of autacoids with vasodilating properties, such as nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarising factor (EDHF), but also with vasoconstricting potential, such as endothelin-1. Corresponding to the physiological situation and vasodilating/ vasoconstricting properties of these endothelial-derived substances, VSMCs respond with changes to their contractile state. Simultaneously, circulating vasoactive substances may also lead to vasoconstriction through direct VSMC activation.8,10,12 In normal coronary vasomotor function, providing an intact endothelial integrity and adequate VSMC reactivity, the net effect of these set of conditions favours vasodilation.
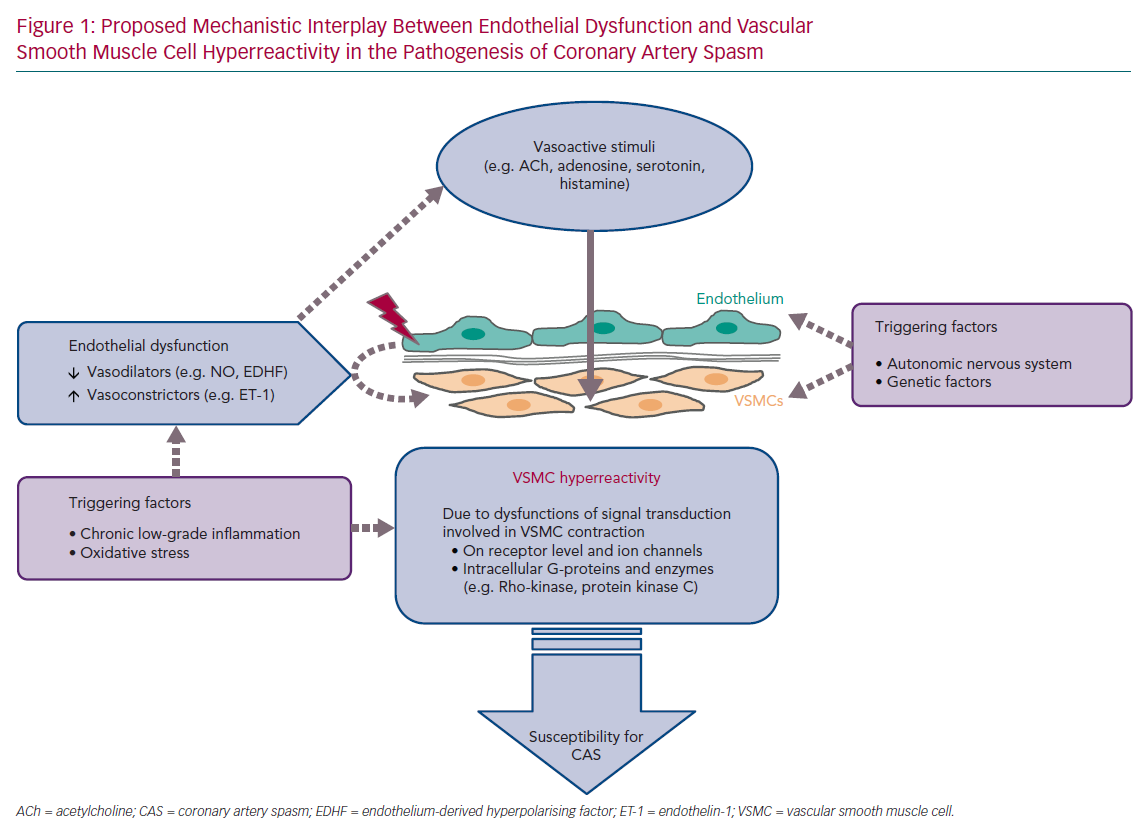
In the presence of impaired endothelial function and/or VSMC hyperreactivity, the net effect of circulating vasoactive substances can be vasoconstriction and this could favour CAS. In 1980, Furchgott and Zawadzki demonstrated in isolated rabbit thoracic aortas that relaxation of arterial VSMCs in response to ACh requires the presence of endothelial cells and that the complete loss of endothelial cells by rubbing off the intimal surface or intraluminal exposure to collagenase resulted in the complete loss of vasorelaxation in response to ACh.13
The link between the increased vasoconstricting potential of coronary arteries in response to ACh and susceptibility to CAS was confirmed by clinical trials.14,15 Shimizu et al. observed vasoconstriction >50% in response to intracoronary ACh infusion in all examined patients with vasospastic angina without epicardial stenosis, while healthy controls showed neither ischaemic ECG changes nor vasoconstriction >50% during ACh provocation testing. In most asymptomatic controls, only mild vasoconstriction (0–25%) or even vasodilation was observed. Vasodilation in response to ACh was more frequent in relatively young controls indicating that age-related physiological impairment of endothelial function may favour the vasoconstrictive potential of the coronary vasculature despite angiographically unobstructed coronary arteries.14 This observation was confirmed by Okumura et al. who also saw a predominantly vasodilatory effect of ACh in young control subjects, whereas patients with CAS showed a significantly and diffusely enhanced vasoconstrictor response to ACh.15 However, these observations do not tell us whether the observed constricting effect of ACh is mainly mediated via a dysfunctional or even absent endothelium or an increased sensitivity of VSMC.
Role of Endothelial Dysfunction in the Pathogenesis of Coronary Artery Spasm
Endothelial dysfunction comprises a spectrum of pathological conditions involving an imbalance between endothelium-derived substances mediating vasodilation, antimitogenic and antithrombogenic properties and substances with vasoconstricting, prothrombotic and proliferative potential.16 In particular, an involvement of NO and endothelin-1 as typical substances synthesised by the endothelium in the pathogenesis of CAS on the epicardial as well as the microvascular level, is supported by various animal and clinical studies. Furthermore, immunohistological findings in endomyocardial biopsies of symptomatic patients with unobstructed coronary arteries revealed an increased expansion of activated endothelial cells, indicating impaired endothelial integrity in patients with microvascular spasm.17
Role of Nitric Oxide
Since NO is one of the most important endothelium-derived vasodilators, impaired NO bioavailability plays a crucial role in endothelial dysfunction. In the setting of CAS, several clinical studies suggested an impaired endothelial NO bioactivity in epicardial coronary arteries of patients with CAS.18,19 Besides, the clinically observed strong vasodilatory effect of the NO donor nitro-glycerine in patients with CAS is also likely to be related to a reduced bioavailability of endogenous NO, which results in a higher basal tone and decreased coronary diameter.15,18 The association of predisposition for CAS with mutations, such as T-786-C, and polymorphisms, such as NOS4a and Glu298Asp, in the endothelial NO-synthase gene (NOS3) further supports the contribution of an impaired endothelial NO bioactivity and endothelial dysfunction in the pathogenesis of CAS.20–2

However, CAS may not only occur at the epicardial but also at the microvascular level. In this regard, experimental ex vivo studies in rat and human arteries indicate the existence of a vessel size depending on the heterogeneity of endothelial vasodilator functions with a predominate contribution of NO to vascular relaxation in relatively large, conduit arteries, such as the aorta and epicardial coronary arteries, while EDHF plays a greater role in resistance arteries, such as the coronary microvessels.11,23,24
Role of Endothelin-1
Endothelin-1 (ET-1), one of the most potent vasoconstrictors, is increased in the coronary and systemic circulation in patients with coronary endothelial dysfunction. Several clinical studies demonstrated higher ET-1 plasma levels in the coronary sinus – baseline as well as during CAS provocation – in patients with a positive epicardial CAS provocation test indicated by vasoconstriction in response to ACh, compared with patients with a normal vasodilatory response.25–27 Although the endothelium is a major source of ET-1, it has been shown that ET-1 is also expressed in macrophages and intimal VSMCs in atherosclerotic tissue specimens from patients who underwent percutaneous revascularisation.28 Furthermore, expression of endothelin-converting enzyme-1 (ECE-1), the key enzyme in ET-1 processing, was ascertained in neointimal VSMCs in rat balloon-denuded arteries as well as in VSMCs and macrophages in human coronary atherosclerotic lesions.29
Experimental and clinical studies provide further evidence that ET-1 is also involved in the pathogenesis of coronary microvascular spasm. In a porcine coronary microvascular spasm model, repeated endothelial denudation of epicardial coronary arteries increased the plasma levels of ET-1 in coronary sinus blood compared with the control group without endothelial denudation, while the chronic administration of an ETA receptor antagonist prevented the coronary microvascular vasoconstrictive response to ACh.30
In a placebo-controlled clinical trial in patients with coronary microvascular dysfunction (defined by a ≤50% increase in coronary blood flow [CBF] in response to the maximal dose of ACh compared with baseline CBF) and non-obstructed CAD, Reriani et al. demonstrated an improvement of microvascular endothelial function after long-term (>6 months) treatment with the ETA receptor antagonist atrasentan.31 Ford et al. used a case-control study to compare peripheral endothelial function and vascular reactivity in patients with epicardial spasm (defined as VSA; vasospastic angina) or coronary microvascular dysfunction (defined as MVA; microvascular angina) with control subjects who had stable chest pain but a normal intracoronary vasoreactivity test result. For the functional ex vivo wire myography-based experiments small resistance arteries (diameter <400 µm) were dissected from gluteal subcutaneous fat biopsies. Reduced vasorelaxation in response to ACh and increased vasoconstrictive response to ET-1 was found in VSA and MVA patients compared with control subjects, indicating a generalised systemic microvascular dysfunction in these patients. This study identifies ET-1 as a potential mediator of endothelial dysfunction and enhanced vasoconstriction in VSA and MVA.32
Role of Vascular Smooth Muscle Cell Hyperreactivity in the Pathogenesis of Coronary Artery Spasm
Vascular tone, defined as the ratio of baseline/maximal vessel diameter, is related to the contractile state of VSMCs. Various mechanisms and stimuli act through activation and inactivation of intracellular pathways involved in the phosphorylation of myosin light chain (MLC) resulting in VSMC contraction. When there is a background of overproduction of contractile stimulators such as ET-1 or absence of relaxing factors such as NO, any additional otherwise innocent contractile stimulus might result in CAS. However, CAS could also be elicited by a low concentration of a contractile stimulus acting on an abnormally sensitive receptor on the VSMCs. Theoretically, dysfunctions in all components of signal transduction involved in the complex regulation of VSMC contraction may contribute to VSMC hyperreactivity, including receptors and ion channels as well as intracellular G-proteins, such as RhoA, and enzymes, such as Rho-kinase and protein kinase C).8,10 A central role of abnormalities located at the level of the VSMCs in the pathogenesis of CAS is also supported by clinical evidence demonstrating that CAS can be provoked by a variety of substances, such as ACh, dopamine, serotonin and histamine, acting directly on VSMCs through different intracellular mechanisms.33–36
Although the cellular and molecular mechanisms triggering VSMC hypercontraction and CAS are still incompletely understood, an increased Rho-kinase activity within the VSMC seems to be substantially involved in the pathogenesis of CAS.37–39 Animal models for CAS demonstrated an upregulation of Rho-kinase in spastic segments of coronary arteries, whereas the Rho-kinase-inhibitor hydroxyfasudil prevented dose-dependently coronary hyperconstrictions.37,38 Fasudil also markedly attenuates ACh-induced coronary vasoconstriction in the clinical setting by preventing the occurrence of chest pain and ischaemic ECG changes in patients with CAS.39 It also ameliorates myocardial ischaemia in patients with microvascular dysfunction.40 However, little is known about mechanisms leading to upregulation and increased activity of Rho-kinase. In this regard, inflammatory mechanisms have been proposed as important contributors causing upregulation of Rho-kinase. In a porcine model chronic focal application of interleukin-1-beta (IL-1-beta), a major inflammatory cytokine, induces coronary intimal lesions at the site of treatment. Focal vasospasm can be provoked at these sites in response to intracoronary serotonin and histamine.41
In further studies using the same porcine model, serotonin-induced hypercontractions at the IL-1-beta-treated site were dose-dependently inhibited by the Rho-kinase inhibitors hydroxyfasudil and Y-27632.37,38 Moreover, upregulation of Rho-kinase expression in the spastic segment seems to be involved in inducing VSMC hypercontraction by inhibition of MLC phosphatase through phosphorylation of the myosin-binding subunit.38 The role of Rho-kinase upregulation by inflammatory pathways is further supported by Hiroki et al. demonstrating that inflammatory stimuli, such as angiotensin II and interleukin-1-beta, upregulate the expression and activation of Rho-kinase in human VSMCs both at RNA- and protein-levels in a time- and concentration-dependent manner. The PKC/NF-κB signalling pathway is apparently involved in the inflammatory stimuli-induced upregulation of Rho-kinase both in vitro and in vivo.42
Endothelial Dysfunction Versus Vascular Smooth Muscle Cell Hyperreactivity
Although there is consistent evidence that endothelial dysfunction is involved in the pathogenesis of CAS, an exclusive role of endothelial dysfunction is challenged by several clinical observations. First, endothelial dysfunction is often associated with common cardiovascular risk factors, such as hypertension, diabetes, dyslipidaemia and atherosclerosis, whereas the prevalence for CAS is comparatively much lower. Thus, it is not surprising that ACh-induced CAS is seen less frequently in patients with chest pain, hypertension and uncontrolled blood pressure or diabetes than in patients without these risk factors.43,44 Moreover, it could be shown by using substance P for the assessment of endothelial-dependent vasodilation, which in contrast to ACh has no direct vasoconstricting action on VSMCs, that endothelium-dependent vasodilation may even be preserved in the spastic segments of epicardial coronary arteries in patients with CAS suggesting that focal vasospasm may result from focal hyperactivity of VSMC.45,46 Clinical findings by Kaski et al. support the hypothesis that focal CAS in patients with variant angina primarily arises from VSMC hyperreactivity to vasoactive stimuli. This study showed that in patients with documented spontaneous CAS, ergonovine-induced CAS was seen at the same site. This indicates that in the presence of a generalised stimulus affecting all coronary arteries, CAS may only occur at a site of local coronary hyperreactivity.47
Distinguishing between endothelial and VSMC dysfunction and the influence on the development of CAS is further hampered by the various angiographic constellations associated with this condition. Most of the findings listed above that indicate a major role for local VSMC abnormalities were obtained in patients with variant angina. This condition is commonly associated with focal sub-occlusive or occlusive spasm.48 However, this angiographic constellation is only infrequently encountered in patients with CAS. When challenged by ACh, a pattern of distally pronounced diffuse spasm of the entire coronary artery is much more commonly encountered.49 The second most frequent finding is microvascular spasm which is characterised by angiographically mild constriction of the epicardial coronary arteries associated with the reproduction of chest pain and often pronounced ST segment depression.50 In the latter two types of spasm we know much less about the respective contribution of abnormalities in endothelial cells and VSMC. Precise characterisation of the clinical picture, the angiographic appearance of the coronary arteries at baseline and the reaction to provocative testing is a cornerstone of understanding the pathophysiology behind CAS. It is likely that the pathophysiology behind CAS is as variable as the clinical presentation and the angiographic patterns.
Role of Chronic Low-grade Inflammation
Accumulating evidence from experimental studies indicate that inflammatory conditions, in particular, via an involvement of Rho-kinase upregulation, may play an important role in the pathogenesis of CAS. An association of CAS with inflammation is also suggested in the clinical setting. Biomarkers of inflammation, such as high-sensitivity C-reactive protein (hs-CRP) and soluble CD40 ligand, are increased in patients with coronary vasospastic angina and unobstructed coronary arteries compared with patients with non-vasospastic angina.51–53 Even minor elevations of hs-CRP serum level are significantly and independently associated with CAS, suggesting that a chronic low-grade inflammatory state may also contribute to CAS susceptibility.52 The observation that cigarette smoking is significantly and independently associated with CAS occurrence and there is an association of increased hs-CRP levels with smoking further supports an involvement of chronic low-grade inflammation in CAS.54
However, inflammation can work as a trigger for both endothelial dysfunction and VSMC hyperreactivity. The exposure of the endothelium to circulating proinflammatory mediators in infective or inflammatory states is associated with impaired endothelial function and inflammation has also been considered as a cardiovascular risk factor.55,56 Elevated CRP levels were identified as a significant independent predictor of a blunted systemic endothelial vasodilator capacity in patients with CAD, while normalisation of CRP levels was associated with a significant improvement of the endothelium-dependent vascular function.57 On the other hand it was shown in an experimental porcine model with chronic IL-1-beta treatment that endothelium-mediated coronary vasodilatation in response to bradykinin, substance P, as well as an increase in coronary blood flow, was preserved at the spastic inflammatory site. Further ex vivo organ chamber experiments confirmed increased serotonin-induced contractions at the IL-1-beta-treated spastic site regardless of the presence or absence of the endothelium, suggesting a primary contribution of VSMC hypercontractions in this model of CAS.58
Based on these observations, it is likely that endothelial dysfunction may provide the background on which additional stimuli manage to produce CAS. Endothelial dysfunction alone could not explain the phasic clinical presentation of patients with vasospasm proven by invasive provocation testing. These patients typically report attacks of resting angina either superimposed on normal exercise tolerance (variant angina) or dyspnoea with exertion, which is more often found in microvascular spasm. Thus, it needs additional triggers, which are not well understood, to explain this pattern of angina in patients with CAS. Endothelial dysfunction, which is a constant condition, alone would probably not explain the sudden occurrence of ≥90% vasoconstriction during coronary spasm provocation test (corresponding to the definition of epicardial CAS according to the Japanese Circulation Society guidelines) with concomitant reproduction of their usual symptoms at home.5 However, the presence of VSMC hyperreactivity coupled with yet to be identified endogenous triggers seems to be sufficient to explain this phasic angina pattern in patients with CAS. The importance of a primary contribution of functional VSMC impairment is further supported by the clinical observation of cases with recurrent epicardial vasospastic angina refractory to nitrates, which would normally be substituted for insufficient production of endothelial NO.59
Conclusion
Due to the heterogeneous clinical presentations of patients with CAS, it is likely that there are several underlying mechanisms involved in its pathogenesis rather than a single mechanism of action. Based on the current knowledge, the presence of VSMC hyperreactivity to vasoactive stimuli seems to be required to explain the phasic clinical pattern seen in patients with CAS. Endothelial dysfunction could provide the background for development of CAS by increasing coronary vascular tone. However, this rather constant milieu provided by the endothelium alone cannot explain the phasic occurrence of angina which is associated with ≥90% vasoconstriction. Abnormalities at the level of the VSMC are also likely to be required to fully explain the clinical picture encountered in patients with the different forms of CAS. Rho-kinase inhibitors, acting directly at the level of the VSMCs by inhibition of Rho-kinase mediated vasoconstriction as well as ETA receptor antagonists, counteracting the increased production of endothelin-1 by dysfunctional endothelial cells, are potential approaches to successfully treat CAS. However, further studies elucidating the underlying pathophysiology of the various forms of CAS are urgently needed before targeted pharmacological therapies can be designed.