







Brugada syndrome was first described in 1993 in a case series of eight patients with recurrent polymorphic ventricular tachycardia (VT) and stereotypical electrographic characteristics in the context of a structurally normal heart.1 Since then, the syndrome has been extensively studied and recognised worldwide as a major cause of sudden cardiac death (SCD) in otherwise healthy patients.2 Recent data support the premise that Brugada syndrome is the most common single underlying aetiology for sudden unexplained death with negative autopsy, representing 28% of cases in the UK.3
Yet more than 30 years since its recognition, there are many questions and uncertainties surrounding the pathophysiology, diagnosis, risk assessment and management of Brugada syndrome. This article aims to examine some of these issues with consideration of recently published data.
Diagnostic Process
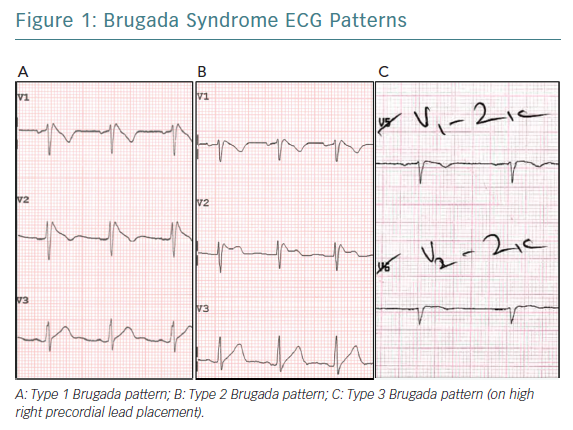
Three distinct ECG patterns have been associated with Brugada syndrome (Figure 1); however, the type 2 and type 3 ECG patterns are less specific for the condition.2,4 The diagnosis of Brugada syndrome requires documentation of the type 1 ECG pattern (consisting of coved-type ST-segment elevation of ≥2mm followed by a negative T wave in the right precordial leads V1 and/or V2) either on a spontaneous ECG or following IV administration of a class I antiarrhythmic agent such as flecainide or ajmaline.2,5
The 2013 diagnostic criteria excluded the requirement for associated clinical features, with diagnosis based purely on the ECG phenotype.5 However, to avoid overdiagnosis of low-risk patients, a further consensus statement on J wave syndrome has proposed the reintroduction of additional clinical evidence to support the diagnosis in patients having type 1 ECG only in the context of sodium channel blockade.4 These clinical criteria include documented VF or polymorphic VT; syncope of probable arrhythmic cause; family history of SCD in a relative younger than 45 years; type 1 ECG in family members; nocturnal agonal respiration; and inducibility of VT/VF at programmed stimulation with one or two premature beats.
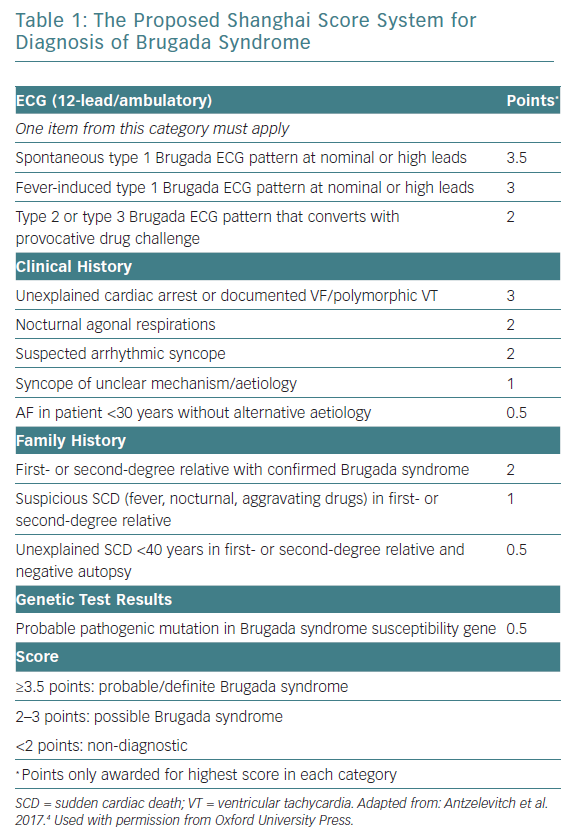
The J wave syndrome consensus group also proposed the novel Shanghai scoring system that takes into account various ECG, clinical history and family history, similar to the Schwartz score used for diagnosis of long QT syndrome (Table 1).5 However, this scoring system is controversial because it was based on expert opinion, rather than appropriate experimental data, and it has not yet been validated.
Past observations have shown that men more commonly present with spontaneous type 1 ECG pattern.1 Cranial displacement of the right precordial ECG leads from the fourth to the second or third intercoastal space – the high right precordial leads – has been shown to increase the sensitivity of the detection of the type 1 ECG pattern, possibly due to better anatomical correlation with the right ventricular outflow tract (RVOT), with no apparent change in prognostic value.2,6,7
In addition, a normal ECG does not rule out Brugada syndrome, as many patients with confirmed diagnosis only have intermittent Brugada ECG patterns.8,9 It has been suggested that prolonged 24-hour ambulatory monitoring using 12-lead ECG with high right precordial lead placement should be used for patients with a normal initial ECG with suspected Brugada syndrome. This may improve diagnostic yield without the risk associated with drug provocation.10,11
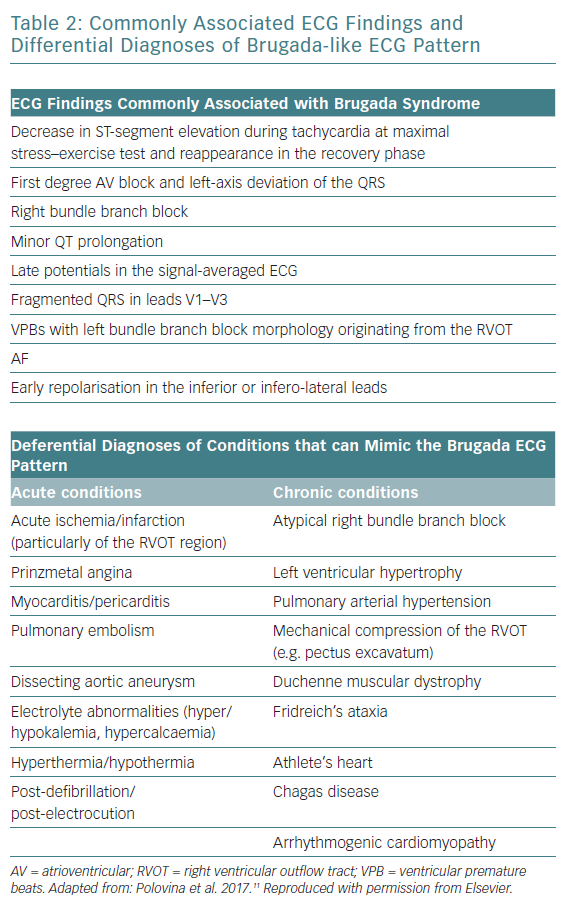
Other ECG findings that are non-diagnostic but are often associated with Brugada syndrome are shown in Table 2. In addition, Brugada syndrome remains a diagnosis of exclusion after considering other differential diagnoses that may chronically or acutely mimic the Brugada ECG pattern (Table 2).12 Normal cardiac structure and function on cardiac imaging is necessary to diagnose Brugada syndrome, since cardiomyopathies are recognised mimics. A reproducible type 1 ECG with sodium channel blockade is helpful to differentiate Brugada syndrome from phenocopies where there is uncertainty over diagnosis after an acute event.
The benefit of using confirmatory diagnostic testing in all patients with non-diagnostic type 2 or 3 Brugada ECG pattern is questionable. Considering the low risk of asymptomatic patients without a spontaneous type 1 ECG, current consensus recommends testing with a class I antiarrhythmic agent to support the diagnosis only if the patient has at least one associated clinical feature.4,13
On the other hand, previous studies have reported worrying false-negative rates on pharmacological challenge testing of symptomatic and asymptomatic patients, with ajmaline being acknowledged as having greatest sensitivity.14,15 It has therefore been suggested that it is worthwhile to repeat a negative pharmacological test using a different agent in cases of high clinical suspicion.12 As with the case of non-pharmacological ECG recording, use of high right precordial lead position during ajmaline challenge testing has been shown to significantly increase the sensitivity of the test.16
Risk Stratification
Symptomatic presentation with previous aborted cardiac arrest or sustained ventricular arrhythmia carries the highest risk of recurrent arrhythmic event in the form of VT/VF, with a previously reported annual rate of about 8%, and secondary prevention with ICD implantation is recommended.5,17 Syncope is a common presentation, occurring in about 30% of patients, and arrhythmia-related syncope is also a well-established risk factor, with an annual event rate of about 2%.11,17 Therefore, it is recommended that a vasovagal cause of syncope is ruled out as these do not seem to carry any additional risk.12,18
Nevertheless, most patients diagnosed with Brugada syndrome are asymptomatic and present a greater challenge for risk stratification and management decisions.12,19 While the established risk of arrhythmic events is much lower for asymptomatic patients at about 1% annually overall, many cases of SCD still occur in this population.12,17,20 Yet despite multiple investigations into predictors of arrhythmic events in asymptomatic patients, no consensus is available for a risk stratification strategy in this population.12
A spontaneous type 1 Brugada ECG pattern is an important established risk factor for cardiac events, which has been reported to confer a three- to fourfold higher risk in asymptomatic patients.21,22 Despite this, only conservative surveillance and risk modification of lifestyle is universally recommended in asymptomatic patients.13 In contrast, a family history of SCD at any age is not an independent prognostic indicator for cardiac events in either symptomatic or asymptomatic patients.19,23
While a multitude of ECG parameters have been associated with increased risk of cardiac arrhythmias, two that have been consistently reported as independent risk factors are abnormal QRS fragmentation (defined as four spikes in one, or eight spikes in all of the leads V1, V2 and V3) and early repolarisation pattern in inferior and/or lateral leads.17,26–29 In addition, AF is more commonly seen in Brugada syndrome than in the general population and has been reported as a risk factor for ventricular arrhythmias in several studies.25,26,30,31
Electrophysiological Study for Risk Stratification
There is ongoing debate regarding the prognostic value of conducting electrophysiological study (EPS) with programmed electrical stimulation for risk stratification in patients without a history of cardiac arrest. Several investigations have demonstrated a positive association between the induction of VT/VF during EPS and the risk of future ventricular arrhythmias, while others have failed to do so.19,21,24,32,33
One important limitation (and potential source of bias) of studies in this area is that patients with a positive EPS are more likely to receive ICD implantation. Therefore, this population may have a higher frequency of recorded ventricular arrhythmias that may not result in cardiac arrest than in patients with negative EPS and no ICD.17,19 Another limitation is the lack of homogeneity between different EPS protocols in the literature.34 While aggressive stimulation protocols with ≥3 extrastimuli are more sensitive, they were found to be less specific than moderate protocols (≤2 extrastimuli).18,35
Despite previous reports describing a strong negative predictive value of EPS in patients without cardiac arrest, a recent systematic review concluded that a negative EPS does not reliably indicate low risk in asymptomatic patients in the presence of other high-risk features (such as spontaneous type 1 ECG, for example).18,34,36 Moreover, the negative predictive value is a function of the low event rate in this low risk population, further limiting the clinical value of EPS.
Gender-specific Risk
Brugada syndrome is about 7–10 times more prevalent in men, and male gender is associated with a higher incidence of ventricular arrhythmias and SCD at diagnosis and follow-up.17,37,38 However, despite the less favourable prognosis generally observed in men, gender alone may not be an independent prognostic indicator and the majority of men remain asymptomatic.17,19
Risk stratification in women with Brugada syndrome has been more challenging as most studies have mainly consisted of male participants. Available data suggest some classical risk factors, such as spontaneous type 1 ECG pattern, are not prognostic in women.38,39
A recent study including 494 women (31% of the total study population) has confirmed the prognostic value of symptomatic presentation with cardiac arrest or syncope in women.40 The only independent risk factors found in asymptomatic women – the majority of patients – were QRS fragmentation and duration >120 ms on ECG. Notably, asymptomatic women without QRS fragmentation had a very low event rate of only 0.1% per year. Previous smaller studies have described the presence of sinus node dysfunction and increased PR interval duration as the best independent markers of cardiac events in asymptomatic women.16,38
Of note is that these gender-specific observations are only relevant for adults (≥18 years), with no significant difference in phenotypic presentation in children.41,42
Genotype and Prognosis
The genetic characterisation of Brugada syndrome has proven to be challenging. The most common and well established Brugada syndrome genotype involve loss of function mutations in the SCN5A gene, representing between 15–30% of diagnosed patients.43 The SCN5A gene encodes for the cardiac voltage-gated sodium channel (Nav1.5) that is activated during the initial rapid depolarisation (phase 0) of the cardiac action potential cycle. About 300 mutations in the SCN5A gene have already been described, yet the role for genetic testing in the diagnosis of Brugada syndrome is limited due to the presence of ‘benign’ SCN5A variants in the general population.4,43
It is clear that the genetic basis for Brugada syndrome is heterogenous and no pathogenic genotype can be currently identified in the majority of patients.12 Moreover, while numerous mutations in several other myocardial genes have also been suggested there is insufficient evidence to establish their unequivocal causality in the pathogenesis of Brugada syndrome.44
Results from several large registry studies have failed to establish an independent association between genotype status and prognosis in Brugada syndrome.12,13 However, recent data suggest that mutations specifically involving the pore region of the SCN5A gene carry a more severe phonotype and were found to be independently associated with the risk of adverse cardiac events in both symptomatic and asymptomatic patients.45
Risk Modification Treatments
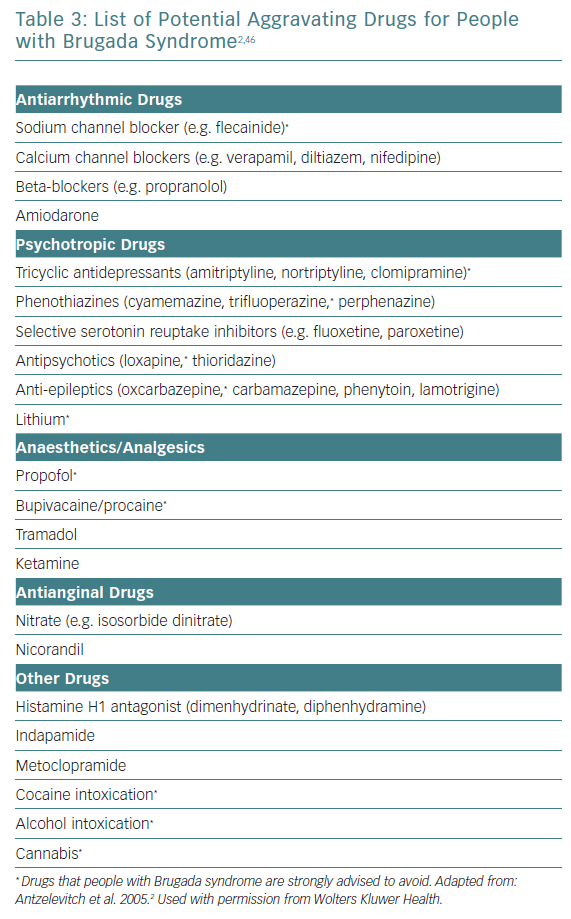
The mainstay of treatment in high-risk patients remains ICD implantation.12 Important risk-reducing strategies for all patients include avoiding excessive alcohol intake, immediate treatment of fever with antipyretics as well as avoidance of potentially aggravating medications (Table 3).5
Quinidine Therapy
Treatment with quinidine can be considered as an adjunct to ICD in patients experiencing electrical storms or frequent appropriate shocks, or as an alternative to ICD in patients with contraindications to implantation. Quinidine reduces the Ito current during epicardial repolarisation and normalises the action potential and prevent re-entry and polymorphic VT formation in experimental models.47
The efficacy of quinidine monotherapy in long-term prevention of malignant ventricular arrhythmias after ICD implantation has been demonstrated in multiple studies.48 A retrospective study showed total elimination of appropriate ICD shocks in 66% (19 of 29) of patients with previous arrhythmic storm or frequent shocks over a mean period of 60 ± 41 months.49 The authors observed a significant and clinically relevant reduction in number of shocks experienced in the remaining patients.
Two main approaches have been previously reported for quinidine monotherapy as an alternative to ICD implantation. The first is guided by the effect of quinidine therapy on inducible VF during EPS. Three long-term prospective studies have reported high rates (76–90%) of prevention of inducible VT during programmed ventricular stimulation while on regular quinidine (600–900 mg daily) for both symptomatic and asymptomatic patients.50–52 No cardiac deaths or definite ventricular arrhythmias were reported while on appropriate quinidine therapy in all patient groups.
The second approach is the empirical use of quinidine for prevention of arrhythmic events without electrophysiological verification. This has so far been mainly evaluated by a randomised trial of quinidine versus placebo of 50 patients with previously implanted ICD.53 While treatment appeared to be effective with no associated arrhythmic events observed, no significant result could be obtained due to low event rate in the placebo group as well as high rates of treatment discontinuation.
One substantial problem with quinidine therapy used as an alternative to ICD implantation is the issue of poor adherence and treatment discontinuation or interruption due to associated adverse effects, most commonly gastrointestinal.48 While treatment with low-dose quinidine (<600 mg daily) is associated with greater tolerability, it has only been investigated in a small number of patients.54,55 Another important, but less common, adverse effect of quinidine is QT interval prolongation that can result in the paradoxical initiation of ventricular arrhythmias.56 These concerns have limited the use of quinidine as a risk modification agent in low-risk asymptomatic people with Brugada syndrome, although an ongoing international registry study hopes to provide evidence to support this (NCT00789165).
Role of Radiofrequency Ablation in Brugada Syndrome
Radiofrequency ablation (RFA) of arrhythmogenic zones in the right ventricular epicardium has emerged over the past decade as a possible future curative treatment option for Brugada syndrome. However, only a small number of studies with limited follow-up periods have reported successful results with RFA in symptomatic Brugada patients.
The first to describe a successful RFA procedure in Brugada were Nademanee et al. using a selected cohort of nine high-risk patients with frequent ICD shocks for ventricular arrhythmias.57 All patients were found to have a unique arrhythmogenic focus at the anterior RVOT on epicardial mapping as well as typical type 1 ECG and inducible VT/VF at baseline. Following ablation, the ECG had normalised in 89% and VT/VF was no longer inducible in 78% of the cohort. Only one of the nine patients had a single subsequent arrhythmic event during the follow-up period (20 ± 6 months).
More recently, Brugada et al. and Pappone et al. described an improved technique for successful elimination of the Brugada syndrome phenotype with epicardial RFA.58,59 The mapping was performed before and after administration of flecainide/ajmaline which resulted in identification of more extensive arrhythmogenic segments in the RV epicardium beyond the RVOT. In the larger and more recent study, the described RFA procedure showed normalisation of ECG and nondeducibility of VT/VF in all of the 135 patients with symptomatic Brugada syndrome and previous ICD.59 Additionally, a type 1 ECG could not be provoked with ajmaline following RFA in the vast majority. During a median follow-up period of 10 months only two patients required a repeat procedure due to recurrent VF.
The only adverse effect reported for all the above studies was mild uncomplicated pericarditis after ablation.58,59 RFA treatment is therefore recommended for symptomatic patients with recurrent ICD shocks or as an alternative to ICD implantation when contraindicated.13,60 Whether this is a suitable alternative to ICD for people with high risk, or even an option for low risk people as a potential ‘cure’ remains to be determined.
Is Brugada Syndrome a Channelopathy or Cardiomyopathy?
Two main pathophysiogical mechanisms have been described for the formation of ventricular tachyarrhythmias leading to SCD in Brugada syndrome. Historically, Brugada syndrome was perceived as a repolarisation disorder, caused by the unequal expression of transient outward potassium current (mediated by a reduction in early sodium inflow) between the epicardium and inner myocardial layers. This results in abbreviation of the epicardial action potential and susceptibility to the formation of re-entry polymorphic ventricular tachycardia triggered by a premature ventricular complex (PVC), due to the epicardial-to-endocardial transmembrane ionic imbalance.61 The evidence for this theory is mainly derived from transmembrane action potential recording in canine right ventricular wedge preparations and is consistent with the clinical effects seen with quinidine, which reduces outward potassium current.62,63
The depolarisation theory is modelled around action potential conduction delay in the RVOT relative to the surrounding myocardium. Under such circumstances, ventricular tachyarrhythmias can be triggered by the resulting unequal membrane potential around the RVOT border, similar to the formation of ventricular tachyarrhythmia seen in circumstances of regional myocardial ischaemia.64 This theory is supported by several clinical studies demonstrating relative conduction delay in the RVOT.65–67
Brugada syndrome was originally described as a disease of cardiac ion channel dysfunction leading to sudden death in otherwise healthy people without the presence of associated structural heart disease.2 However, evidence demonstrating normalisation of the pathognomonic ECG pattern and elimination of the arrhythmic disposition in most patients following radiofrequency ablation of the RVOT epicardium supports the theory that structural abnormalities play an important role in the pathophysiology of Brugada syndrome.68
Indeed, recent results suggest that microanatomical changes such as increased collagen, fibrosis and reduced gap junction expression, which may be mediated by underlying pan-myocardial inflammation, are responsible for the characteristic electrographic pattern and arrhythmic susceptibility.68,69
As a result of these findings, Brugada syndrome and arrhythmogenic cardiomyopathy have been proposed to be part of the same disease spectrum.70 Although these two conditions have distinct macropathological appearance, known genetic predisposition and clinical features, several overlapping manifestations can be seen.2 The Brugada ECG pattern is seen in some patients with arrhythmogenic cardiomyopathy and sudden death can occur in the initial phase of the condition in the absence of its characteristic structural changes.71 Experimental data suggest that the arrhythmogenic process in both conditions is mediated by dysfunction of a complex protein network in the myocardial intercalated disc microarchitecture called the connexome.71 Nevertheless, while dysfunction of the connexome process may represent a common aetiology for Brugada syndrome and arrhythmogenic cardiomyopathy, the phenotypic presentation in Brugada syndrome appears to be restricted to microanatomical changes and sodium channel dysfunction in the RVOT region.
It therefore appears that the pathophysiology of disease generation and progression is more complex than initially described and greater understanding of these underlying processes is likely to improve the diagnosis and management of Brugada syndrome.
Conclusion
Brugada syndrome has been clouded in controversy since its first description more than three decades ago. While expert consensus has been reached on many aspects of the disease, a significant number of core issues such as the underlying pathophysiology of the disease, diagnostic criteria and risk stratification of asymptomatic patients remain unresolved. The unravelling of its underlying pathological mechanisms coupled with improved techniques and protocols for invasive mapping and ablation procedures shine an optimistic light on the possibility of a future cure for the syndrome.