








We are witnessing an epidemic global increase in the prevalence of obesity and its clinical consequences (e.g. insulin resistance and diabetes). This epidemic has been potentiated and sustained by the widespread adoption of unhealthy lifestyles in broad swathes of the population and is characterised by a sedentary lifestyle and an imbalance between the type and characteristics of nutrition, dominated by an excess of calorie intake. Its effects have come to offset the decline in cardiovascular (CV) mortality achieved in recent years as a result of marked therapeutic advances.1,2
The prevalence of atherogenic dyslipidaemia (AD) has increased considerably. AD is characterised by the coexistence of profound qualitative and quantitative modifications in lipid metabolism. The excess of non-LDL particles is a distinctive feature, with an increase in triglyceride (TG)-rich lipoproteins (TRL), low HDL cholesterol levels, accumulation of lipoprotein remnants (i.e. small very LDL [VLDL] and intermediate-density lipoprotein [IDL]), a preponderance of numerous small and dense (sd) LDL particles and postprandial hyperlipidaemia.3–5
The management of AD requires therapeutic lifestyle modification coupled with pharmacological intervention aimed at reducing CV risk. Because of the range of cardiometabolic alterations these patients present with, trying to attenuate their CV risk can pose formidable management issues.6,7
Atherogenic Dyslipidaemia and its Relationship with Atherosclerotic Cardiovascular Disease
The increasing prevalence of obesity is directly associated with the increase in type 2 diabetes (T2D) and metabolic syndrome (MS), which, in turn, are associated with lipoprotein abnormalities described as AD. AD is causally linked to the development and progression of atherosclerotic CV disease (ASCVD).8,9 The relationship between AD and ASCVD is supported by prospective longitudinal cohorts, clinical evidence and genetic linkage studies. As an example, the best predictor of risk of MI at the population level in the INTERHEART study was the apolipoprotein (apo) B100/apoA-I ratio, reflecting the correlation between all apoB (atherogenic lipoproteins) and HDL (representing the classically anti-atherogenic particles).10 In addition, a huge registry of almost 140,000 patients hospitalised in the US due to acute coronary syndromes (ACS) showed that more than half had LDL cholesterol levels <2.59 mmol/l, whereas mean HDL cholesterol and TG values were <1.03 and >1.81 mmol/l, respectively.11,12 In these patients the LDL cholesterol level was not reflecting the real burden of atherogenic lipoproteins; this was more aptly quantified by non-HDL cholesterol (total cholesterol minus HDL cholesterol), a better predictor of CV risk in these individuals.
The evidence goes beyond epidemiological studies. The relationship between AD and ASCVD has also been demonstrated in prospective randomised clinical trials using statins. Even when treated with statins, patients with the AD phenotype have a higher risk of CV events than those without AD.13,14
The Pravastatin or Atorvastatin Evaluation and Infection Therapy – Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial showed that among patients receiving high-intensity statins after an ACS, those with TG <1.69 mmol/l (adjusted by HDL cholesterol and LDL cholesterol levels) had a lower risk of coronary events (HR 0.80; 95% CI [0.66–0.97]; p=0.025) than those with TG exceeding this threshold.15
Similarly, in the Incremental Decrease in End Points through Aggressive Lipid Lowering (IDEAL) and Treating to New Targets (TNT) studies, even in patients who reached LDL cholesterol <1.81 mmol/l, the risk increased 63% (p<0.001) when comparing the highest quintile of TG levels with the lowest one.16 Long-term (>20 years) follow-up of the Bezafibrate Infarction Prevention (BIP) study showed a significant association between elevated TG and all-cause mortality.17 In addition, in a meta-analysis of prospective studies in patients treated with statins, increased TG concentrations were independently correlated with coronary disease and predicted recurrent ischaemic events in patients with a history of ACS treated with statins.11,18,19
Some studies have shown that the association between plasma TG concentrations and CV risk is attenuated when adjusted for other lipid parameters. In a meta-analysis by the Emerging Risk Factors Collaboration, which included data from 302,430 individuals, there was a significant association between TG concentrations and CV risk, but this association was attenuated after adjusting for HDL cholesterol and non-HDL cholesterol.20 In some respects, the case can be made that because non-HDL cholesterol includes lipoproteins that can carry TGs, this likely represents an ‘over-adjustment’ of two highly inter-related risk factor covariates. However, in a more recent analysis, among approximately 46,000 high-risk patients on statin therapy whose LDL cholesterol was well controlled, TG >169 mmol/l was independently and significantly correlated with CV events even after adjusting for LDL cholesterol, non-HDL cholesterol and HDL cholesterol.21
TRL particles (precursors of LDL, including small VLDL and IDL) can be estimated in clinical practice as total cholesterol minus LDL cholesterol minus HDL cholesterol. TRLs are associated with increased CV risk.22,23 Varbo et al. showed that each 1 mmol/l increase in TRLs is associated with a 2.8-fold increase in CV risk independent of the HDL cholesterol level.24 Directly measured TRLs were correlated with an increased risk for ASCVD events in both the Framingham Heart Study and the Jackson Heart Study.25 TRLs are also potently proinflammatory, which likely contributes to their overall atherogenic profile.26 Highlighting the importance of TRLs, postprandial TG concentrations are a stronger predictor of CV events than fasting TG levels. Although most individuals are in a postprandial state during most hours of the day, changes in postprandial TG levels can have a significant effect on the development of atherosclerosis.27–29
In summary, there is convincing evidence that AD is highly atherogenic, although the true significance of each component in this context is incompletely characterised and understood. Human genetic evidence suggests that TRLs contribute causally to the development of ASCVD.12 Gene variants leading to higher levels of plasma apoB-containing lipoproteins, including TRL, consistently increase ASCVD risk.30
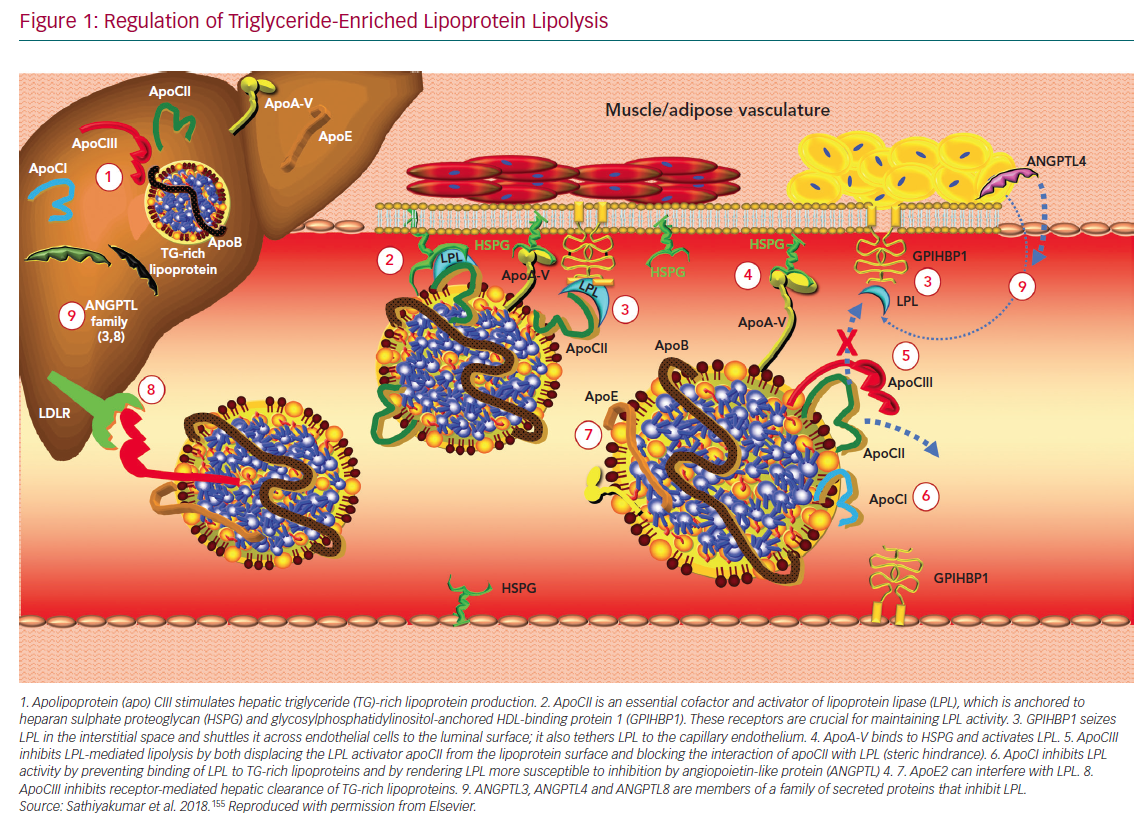
Lipoprotein lipase (LPL) plays a critical role in the disposal of TGs carried with chylomicrons and apoB100-containing lipoproteins. LPL is tethered to vascular endothelial cells via glycosylphosphatidylinositol-anchored HDL-binding protein 1 and hydrolyses TGs within the core of TRLs (Figure 1).
LPL activity can be regulated by changes in nuclear expression of the gene for LPL, but it is also responsive to a variety of effector molecules. LPL is inhibited by apoCIII and activated by apoCII and apoA-V.31 Angiopoietin-like protein (ANGPTL) 3 and ANGPTL4 exert inhibitory effects via distinct mechanisms. ANGPTL3 stimulates cleavage of LPL from GP1HBP by proprotein convertase subtilisin/kexin types 3 and 6, rendering it inactive.32 LPL monomers are catalytically inactive; the active form is an LPL dimer. In contrast, ANGPTL4 competitively inhibits LPL by inducing the dissociation of its constituent dimers.33 The severity of diabetes can also affect the dimeric integrity of this enzyme.34
The genetics of LPL and the effector molecules that regulate its activity support the conclusion that TGs are an independent risk factor for ASCVD. Loss-of-function mutations in apoCIII result in lower mean serum TG levels than in patients who express normal levels of this enzyme that are correlated with significant reductions in ASCVD risk.35,36 Similarly, a variety of genetic polymorphisms giving rise to reduced activity of ANGPTL3 and ANGPTL4 result in lower mean TG levels and lower risk for ASCVD compared with wild-type controls.37–39 Patients with loss-of-function mutations in apoA-V have higher TG concentrations and augmented risk for ASCVD and ischaemic stroke.40,41 Consistent with these changes, gain-of-function mutations and loss-of-function mutations in LPL are correlated with lower TGs/lower ASCVD risk and higher TGs/higher ASCVD risk, respectively.35
Epidemiological data associate low HDL cholesterol with heightened ASCVD risk, although a more recent analysis has questioned this.19,42-44 It was long believed that treating low serum HDL cholesterol concentrations would reduce residual risk. However, clinical outcomes trials targeting low HDL cholesterol with different pharmacological interventions failed to reduce CV endpoints, and, similarly, genetic studies do not support a protective role of HDL cholesterol in humans.45-49 Together, these findings imply that HDL cholesterol may be considered a metabolic marker of increased CV risk rather than a therapeutic target. It will be some time before the HDL proteome and lipidome are understood well enough to tailor therapeutic interventions that affect ASCVD risk.50,51
The Study to Investigate CSL112 in Subjects With Acute Coronary Syndrome (AEGIS-II; NCT03473223) is a large Phase III trial testing the capacity of an infusible human apoAI preparation to reduce the risk of CV events in patients with a history of ACS. The trial will also evaluate the efficacy of CSL112 (apoA-I [human]) in inducing atherosclerotic plaque regression by promoting reverse cholesterol transport.
Abnormalities in Lipid Metabolism: The Perfect Storm
The aforementioned phenotypic characteristics of AD reflect an abnormal metabolism of TRL, conditioned by both genetic and acquired factors that also affect HDL and LDL particles.52 In the setting of insulin resistance, insulin has reduced capacity to inhibit hormone-sensitive lipase in adipose tissue. This leads to a constitutive release of fatty acid from visceral adipose tissue stores. The liver can dispose of this fatty acid in multiple ways:
- It can be oxidised in the mitochondrial matrix.
- It can be reassimilated into TG and secreted in VLDL particles.
- Some can be shunted toward gluconeogenesis by activation of phosphoenolpyruvate carboxykinase, which will exacerbate the hyperglycaemia of insulin resistance.
- If all pathways become saturated, excess TG will be stored in the liver and manifest as hepatic steatosis.
In addition to the overproduction and increased hepatic secretion of VLDLs, there is reduced capacity to lipolyse TRLs because of decreased LPL activity. Insulin resistance can reduce nuclear expression of LPL, leading to increased production of apoCIII and decreased production of apoCII.53 Secondary to both hepatic overproduction of TRL and their reduced catabolism, there is a considerable increase in TRLs in the serum.
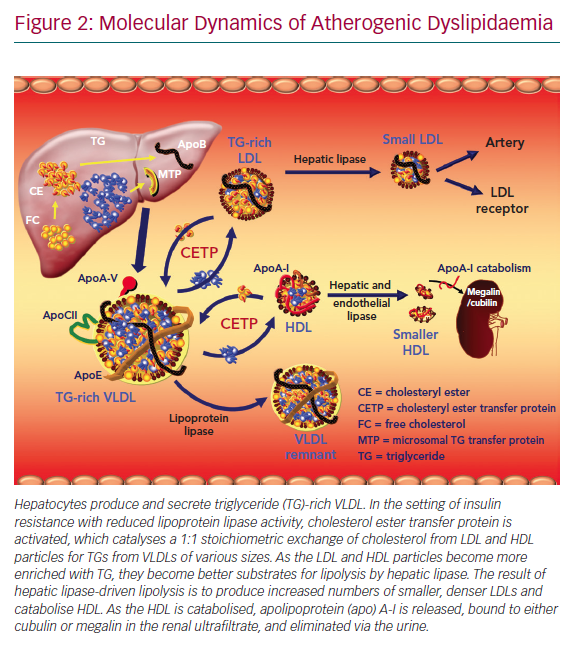
As TRLs accumulate in serum, the activity of cholesterol ester transfer protein (CETP) increases. CETP catalyses the neutral lipid of exchange of TG out of TRLs for cholesterol from both HDLs and LDLs (Figure 2).54 As the HDLs and LDLs become more enriched with TG, they become better substrates for lipolysis by hepatic lipase. Hepatic lipase catabolises HDL and promotes the wasting of apoA-I by the kidney. Hepatic lipase also converts large, buoyant LDLs into smaller (sdLDL) and more numerous ones, rendering the LDL fraction more atherogenic.55 HDL cholesterol levels decrease via other mechanisms as well. There is an insulin response element in the gene for apoA-I, the primary apolipoprotein constituent of HDL particles.56,57 As the liver becomes more insulin resistant, less apoA-I is produced and there is less HDL biogenesis. Adipocytes express the ATP-binding membrane cassette transport protein A1 (ABCA1). Insulin resistance downregulates expression of ABCA1 on the surface of adipocytes and reduces HDL formation by these cells.58–60 Chylomicrons are enriched with apoA-I. Insulin resistance reduces the release of this apoA-I in serum by inhibiting LPL. In addition, within the milieu of insulin resistance or diabetes, HDL particle concentrations are not only quantitatively reduced, but also tend to be dysfunctional and thus are not able to perform their primary functions, including reverse cholesterol transport and inhibition of oxidative and inflammatory phenomena.61
The sdLDL particles are highly atherogenic.62 There is accumulating evidence that smaller LDL particles are more atherogenic than larger, more buoyant ones. In addition, sdLDL:
- Is more susceptible to oxidation (oxidised LDL is avidly scavenged by activated macrophages in the subendothelial space, giving rise to foam cell cells);63
- Contains apoB100, which undergoes conformational alteration as the particle decreases in volume and size, resulting in lower affinity for, and clearance by, the LDL receptor;64 and
- Has increased affinity for proteoglycans in the subendothelial space, exacerbating lipoprotein retention.5
An increase in the hepatic content of TG also promotes an increase in the size of the TRL pool and augmentation of VLDL biosynthesis and secretion. All these observations help explain the extensive and more premature development of ASCVD in patients with insulin resistance and AD.65–67
In summary, changes in lipid metabolism in the setting of insulin resistance or diabetes are profound and include an excess of apoB, including remnants, HDL, which is decreased in quantity and function, and changes in LDL size and particle number. These metabolic changes, coupled with the pro-oxidative, proinflammatory and prothrombotic state of insulin resistance, predispose to the development of accelerated atherogenesis in ‘cardiometabolic’ patients.
Apolipoprotein B and Non-HDL Cholesterol as Risk Markers
ApoB measurement represents an estimation of all atherogenic lipoproteins, namely VLDL, IDL, LDL and lipoprotein(a) (Lp(a)), because they all contain a molecule of apoB100. In the same way, each chylomicron particle or its remnants contain a molecule of apoB48 (a truncated form of apoB100). In AD there is overproduction not only of VLDL, but also of apo B. In patients with cardiometabolic risk, the total number of sdLDL particles is increased, and hence apoB is elevated. Neither the sdLDL concentration nor the apoB level is reflected in LDL cholesterol measurements.68 Among patients with insulin resistance, LDL cholesterol is frequently low or ‘normal’, despite increases in apoB. Non-HDL cholesterol is a simple and practical calculation (total cholesterol minus HDL cholesterol) that represents an estimation of the cholesterol concentration within all atherogenic lipoproteins. It is defined as a secondary treatment target in most international dyslipidaemia guidelines.69–72 Using sophisticated techniques, the number of particles of each lipoprotein class and its subclasses can be quantified, but its application in daily clinical practice is an area of continuing investigation.73
Non-HDL cholesterol and apoB are better therapeutic targets in patients with AD.74 In that sense, most guidelines established for patients at very high risk, include objectives of LDL cholesterol <1.81 mmol/l, non-HDL cholesterol <2.59 mmol/l and apoB <1.56 µmol/l, and in those at high risk of LDL cholesterol <2.59 mmol/l, non-HDL cholesterol <3.36 mmol/l and apoB <1.75 µmol/l. In addition, therapeutic effort should be made to reduce TG burden in serum through lifestyle modification and medication as indicated.75
Non-pharmacological Interventions: Therapeutic Changes in Lifestyle
Healthy Eating
Favourable therapeutic changes in lifestyle constitute the basic approach and the cornerstone of AD treatment. The greatest benefit is obtained with a reduction in saturated and trans-fats intake, along with an increase in consumption of mono- and polyunsaturated fats. It is essential to reduce the excess of carbohydrates in the diet, especially refined sugars.76,77 The Mediterranean diet seems to be more effective than a low-fat diet; the Mediterranean diet has been shown to significantly reduce the total cholesterol:HDL cholesterol ratio and non-HDL cholesterol, and to reduce clinical events and CV mortality.78,79
Both low-fat and low-carbohydrate diets affect lipid levels. The low-fat diet has little effect in decreasing total cholesterol and LDL cholesterol, whereas the low-carbohydrate diet shows more favourable effects on TG and HDL cholesterol. In addition, the consumption of sea fish or omega-3 fatty acids has favourable effects.
In conclusion, lowering the dietary carbohydrate content or losing weight appears to attenuate AD, whereas reducing the total fat or saturated fat content has little effect.80 Weight loss is associated with significant relief of insulin resistance.
Regular Physical Activity
The effects of physical activity on serum lipids have been widely studied. Regular aerobic exercise is associated with increased skeletal muscle and systemic tissue insulin sensitisation.81,82 As this occurs, HDL cholesterol tends to increase and TGs and sdLDL decrease.83,84 Regular physical activity is a key recommendation in the approach to AD.85
Unfortunately, despite the benefits, long-term adherence to lifestyle changes is often difficult to sustain over time.86
Current Pharmacological Interventions
Statins
Statins are first-line drugs in the treatment of AD. They comprise a pharmacological group that inhibit the rate-limiting step of cholesterol biosynthesis catalysed by 3¢-hydroxy-3¢-methylglutaryl coenzyme A. By decreasing intrahepatocyte concentrations of cholesterol, the statins activate the nuclear transcription factor sterol regulatory element binding protein-1c, which increases the cell surface expression of LDL receptors (LDLR). Thus, statins reduce circulating levels of LDL cholesterol by:
- decreasing cholesterol biosynthesis and VLDL secretion; and
- increasing the clearance of LDL particles from the circulation.
In addition, statins can reduce plasma TGs by 15–20% and increase HDL cholesterol by up to 15%.87,88 However, the effect of statins on sdLDL has not been completely clarified.89 A recent meta-analysis of six statin trials, including 802 subjects, demonstrated that statins significantly reduce circulating levels of apoCIII, which likely contributes to their modest TG-lowering capacity.90 In addition to their lipid-lowering effects, the statins exert cholesterol-independent pleiotropic actions, which have been widely studied and contribute to the stabilisation of atherosclerotic plaques and reverse endothelial dysfunction, among other effects.91 The beneficial effects of statins have been extensively demonstrated in both primary and secondary prevention studies because they incontrovertibly reduce risk for MI, ischemic stroke, revascularisation by both percutaneous transluminal coronary angioplasty and coronary artery bypass grafting, as well as cardiovascular and all-cause mortality.92,93
There is some concern that treatment of dyslipidaemia with statins increases the incidence of diabetes. As first reported by Ridker et al. in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER), rosuvastatin therapy is associated with an increased risk for new-onset diabetes.94 A subsequent meta-analysis showed that statins increase the risk for diabetes by approximately 9%.95 However, this does require some contextualisation. As shown in JUPITER, compared with placebo, statin therapy accelerates time to onset of diabetes by only 5.4 weeks and, for those with risk factors for diabetes, 134 vascular events or deaths were prevented for every 54 new cases of diabetes diagnosed.96 Thus, the benefit:risk ratio of statin therapy is quite favourable.
Subsequent work showed that the greater the number of components of MS a patient has, the higher the risk for statin-induced diabetes.97 In general, patients with MS have the highest risk for statin-accelerated diabetes. The mechanism(s) for this are as yet unknown. With low-dose statin therapy 1,000 patients have to be treated for 1 year in order to see one new case of diabetes; with moderate- to high-dose statin therapy, 500 patients have to be treated for 1 year to see one new case of diabetes.98 Statin therapy should not be withheld out of concern that it may precipitate diabetes; it has been shown that diabetics derive every bit as much benefit from statin therapy as do non-diabetics.99 Moreover, it has been suggested that patients with features of MS may derive particular benefit from statin therapy.100
Fibrates
Fibrates are weak agonists of the nuclear transcription factor peroxisome proliferator-activated receptor (PPAR) alpha and regulate the expression of genes that influence lipid metabolism. Fibrates were found to improve glucose homeostasis via activation of PPAR-alpha and increases in adiponectin levels.101 Fibrates lower TGs by 25–50%, increase HDL cholesterol 5–30%, reduce LDL cholesterol up to 20% and decrease sdLDL particles, postprandial TGs and TRLs.102,103
In both the Helsinki Heart Study (primary prevention) and the Veterans Administration Low HDL Intervention Trial (secondary prevention), gemfibrozil significantly reduced risk of the primary composite endpoint.104,105 However, because gemfibrozil reduces statin glucuronidation and can potentiate the risk of rhabdomyolysis, gemfibrozil should not be used in combination with a statin.106,107 Fenofibrate is substantially safer than gemfibrozil when combined with a statin.108 Fenofibrate therapy has been evaluated in two prospective randomised studies of patients with diabetes.
In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, fenofibrate as monotherapy failed to reduce the primary composite endpoint, but did reduce the risk for MI, stroke, revascularisation and multiple microangiopathic endpoints, including retinopathy, nephropathy and neuropathy.109 In a subgroup analysis of patients with TGs >2.26 mmol/l and HDL cholesterol <1.03 mmol/l, fenofibrate significantly reduced the primary composite endpoint by 27%. Although fenofibrate may increase serum creatinine concentrations, it has actually been shown to be nephroprotective and does not adversely affect renal function.110,111 The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial also failed to meet its primary endpoint in patients on statins randomised to either fenofibrate or placebo.112 However, at baseline, both HDL cholesterol and TG levels were very near normal. In meta-analyses, fibrates show a reduction in CV morbidity and mortality, but not in total mortality.113,114 In another meta-analysis, fibrates decreased major CV events by 13%, but this benefit was evident only in subjects with increased TG (>2.26 mmol/l).115 It is in the setting of AD where fibrates are most effective.
As fibrates are weak PPAR-alpha agonists with limited efficacy and dose-related adverse events, a new generation of highly specific PPAR-alpha agonists, known as selective PPAR-alpha modulators, have been developed that preserve the beneficial effects of fibrates and eliminate the unwanted side effects. Recently, pemafibrate was introduced; it is >2,500-fold more potent than fibric acid, with a greater lipid-modifying efficacy and an improved safety and tolerability profile.116 Pemafibrate is being evaluated in the CV outcomes trial, Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT), studying a high-risk diabetic population on statin therapy.117
Cholesterol Absorption Inhibition: Ezetimibe
Ezetimibe reduces the absorption of dietary and biliary cholesterol along the brush border of jejunal enterocytes. Ezetimibe inhibits the transmembrane sterol transporter Niemann–Pick C1-like protein.118 This reduces the amount of cholesterol delivered to the liver and stimulates increased expression of LDLR along the hepatocyte cell surface.119 As monotherapy, ezetimibe reduces LDL cholesterol by approximately 15–20%, but it is most commonly used in combination with a statin, where it provides additive benefit for reducing LDL cholesterol, non-HDL cholesterol and apoB and is safe.120–122
Recently, the IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE IT) demonstrated that ezetimibe provides an incremental reduction in CV events among patients with a previous ACS and being treated with a statin.123 It has been successfully used in patients who do not tolerate statins and in those who are not at goal with adequate doses of statins.119 Ezetimibe can be combined with any statin, at the usual single dose of 10 mg/day, with no significant side effects reported, except for occasionally mild elevation of liver enzymes or myalgia.
In a recent meta-analysis of eight studies including 80,790 diabetics and 85,555 non-diabetics, with a mean follow-up of 45 months, the risk for ASCVD events was significantly less with ezetimibe–statin combination therapy than statin monotherapy in both diabetic (relative risk 0.69; 95% CI [0.67–0.73]; p<0.00001) and non-diabetic (RR 0.68; 95% CI [0.52–0.90]; p=0.006) subjects.124 In a post hoc analysis of IMPROVE IT, the diabetic subgroup on ezetimibe plus simvastatin achieved a significantly lower mean LDL cholesterol than the group on placebo plus simvastatin (1.27 versus 1.73 mmol/l, respectively; p<0.001), and the RR reduction for MI and stroke in diabetics was 24% and 39%, respectively. Thus, the benefit of adding ezetimibe to statin appeared to be enhanced in patients with diabetes.125
Omega-3 Fatty Acids
The omega-3 fatty acids (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) are long-chain polyunsaturated fatty acids that inhibit the synthesis of VLDL and TG in the liver. Although the combination of EPA and DHA reduces serum TGs and VLDL, it also induces an elevation in LDL cholesterol that is proportional to the baseline TG level.126,127 No recent study in the statin era has been able to demonstrate CV risk reduction using combinations of EPA and DHA.128 The Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH) trial is currently testing whether or not a combination of EPA and DHA will affect the risk for ASCVD events in high-risk diabetic patients on a statin background.
The Japan EPA Lipid Intervention Study (JELIS) treated Japanese patients on low-dose statin therapy with 1.8 g EPA or placebo.129 The addition of EPA did provide incremental risk addition beyond statin therapy, but patients were not blinded to therapy and the doses of statin used were quite low (oral simvastatin 5 mg once daily, or oral pravastatin 10 mg, once daily). In addition, the only endpoint that achieved statistical significance was unstable angina requiring hospitalisation. JELIS did not achieve significant reductions in endpoints such as non-fatal MI or stroke, CV mortality or need for revascularisation. Hence, given these limitations, this approach did not become very widely adopted.
Recently, the Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial (REDUCE-IT) showed that treatment of high-risk individuals (58% with diabetes) with 4 g EPA ethyl ester daily (mean TG 2.40 mmol/l, mean LDL cholesterol 1.68 mmol/l) resulted in relative reductions of 25% in the incidence of major adverse cardiac events (MACE) and 20% in CV mortality against a statin background. Entry criteria for the REDUCE-IT trial included age >45 years with established CV disease or age >50 years with diabetes and one or more additional risk factor, fasting TG 1.69–5.63 mmol/l, LDL cholesterol 1.06–2.59 mmol/l and a stable dose of statin for ≥4 weeks. This benefit was independent of both baseline TG and LDL cholesterol.130 The precise mechanisms by which EPA exerts these benefits remains under active investigation, although it is likely that the inhibition of oxidative processes and the augmented production of downstream effectors such as resolvins and protectins play beneficial roles.131
Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors
Evolocumab and alirocumab, monoclonal antibodies (mAbs) that inhibit proprotein convertase subtilisin/kexin type 9 (PCSK9), have been approved for use and are available in a number of countries. They are indicated for use with or without combination statin therapy to reduce LDL cholesterol in patients with established ASCVD or heterozygous familial hypercholesterolaemia. PCSK9 regulates the expression of LDLRs by shuttling LDLR into the lysosome for proteolytic destruction.132 Hence, when plasma PCSK9 is increased or exhibits augmented functionality, the density of hepatocyte surface LDLR is reduced, plasma LDL cholesterol increases and the risk for ASCVD increases.133
In contrast, when PCSK9 is inhibited by mAbs directed against it, cell surface expression of LDLR is increased and plasma LDL cholesterol decreases due to increased clearance.134,135 The inhibition of PCSK9 with mAbs results in a profound decrease in LDL cholesterol of approximately 50–70%. These agents also induce significant reductions in non-HDL cholesterol, apoB and Lp(a).136 The magnitude of the reduction in Lp(a) with the PCSK9 mAbs is proportional to the baseline level.137,138 Patients with low Lp(a) typically experience no to small changes in this lipoprotein. The mechanism(s) by which these agents reduce Lp(a) is a matter of intense debate.139,140 The PCSK9 mAbs are available for biweekly or monthly subcutaneous injection and have a favourable safety profile.
Two landmark studies have been conducted with CV endpoints including thousands of patients randomised to a PCSK9 inhibitor or placebo against statin backgrounds. The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) study randomised 27,564 patients with established ASCVD to either evolocumab or placebo against a statin background. LDL cholesterol decreased 59% (mean level achieved in the active treatment group: 0.78 mmol/l). The risk for the primary (HR 0.85; 95% CI [0.79–0.92]; p<0.001) and secondary composite endpoints 20% (HR 0.80; 95% CI [0.73–0.88]; p<0.001) were substantially reduced after an average follow-up of 2.2 years.141 Evolocumab significantly reduced CV risk in patients with diabetes and did not increase the risk of new-onset diabetes.142 Evolocumab therapy was not correlated with an increased risk for cognitive impairment even when LDL cholesterol was reduced to less than 0.26 mmol/l.143
The Odyssey Outcomes study included 18,924 patients who were between 1 and 12 months after an ACS; 89% were treated with high-intensity statins but did not reach their LDL cholesterol goal (<1.81 mmol/l) and mean baseline LDL cholesterol was 2.25 mmol/l. Patients were randomised to alirocumab 75/150 mg twice weekly versus placebo.144 LDL cholesterol decreased 54.7% in those treated with alirocumab, and the mean follow-up was 2.8 years. The primary endpoint, a combination of coronary death, non-fatal MI, fatal or non-fatal stroke and hospitalisation for unstable angina, decreased 15% (HR 0.85; 95% CI [0.78–0.93]; p=0.0003). Of great interest in this trial was the observation that alirocumab reduced the risk for the primary composite endpoint significantly more in diabetic patients than in patients with prediabetes or those who were normoglycaemic (2.3% absolute risk reduction versus 1.2% and 1.2%, respectively).145
In a variety of patient types with AD, alirocumab and evolocumab demonstrate excellent capacity on top of statins for reducing atherogenic lipoproteins other than LDL cholesterol.146–148 Both evolocumab and alirocumab reduce serum concentrations of VLDL, remnant lipoproteins and LDL particle numbers.149,150 In the FOURIER trial, the reduction in Lp(a) potentiated by evolocumab provided incremental risk reduction over and above LDL cholesterol reduction, an important advance in dyslipidaemia management.137 Although a Mendelian randomisation study suggested that there may be a signal for increased risk of diabetes with a PCSK9 mAb, to date there is no evidence that either evolocumab or alirocumab increases the risk for impaired glucose tolerance, impaired fasting glucose or diabetes.151–154
Conclusion
Despite statin therapy, among patients with AD LDL cholesterol reduction is inadequate because significant residual risk remains. At least some of this risk is attributable to inadequate LDL cholesterol reduction. Clearly, however, mounting evidence suggests that inadequate reduction of other apoB-containing lipoproteins also contributes to residual risk. In the setting of insulin resistance, patients experience a large increase in apoB-containing lipoproteins characterised by elevated levels of VLDL, TRLs and LDL particles. These changes occur in response to increased hepatic TG and VLDL production, inhibition of LPL, activation of CETP and hepatic lipase and reduced clearance of apoB-containing lipoproteins. AD is also characterised by substantial changes in HDL metabolism at the level of multiple organs and cell types. The contributions of low HDL, HDL dysfunction and impaired reverse cholesterol transport to the net effect of AD are matters of continuing investigation. In order to better reduce residual risk among patients with AD and TGs >2.26 mmol/l, it is generally accepted that non-HDL cholesterol and apoB are better targets of therapy, and their aggressive reduction is a clinical priority.
Among the established therapies, statins are the first-line treatment. Ezetimibe and the PCSK9 mAbs constitute important additional therapies that should be used in patients who fail to reach their LDL cholesterol goal with statins alone or in statin-intolerant subjects. Fenofibrate can be considered in patients with AD, especially in those with high TG and low HDL cholesterol despite the use of statins in adequate doses. Although studies with CV outcomes have been inconsistent in demonstrating benefits with fibrates, post hoc analyses of the subgroups with AD (high TG and low HDL) consistently demonstrate ASCVD risk reduction. Based on the REDUCE-IT trial, EPA monotherapy constitutes an exciting, highly efficacious approach to reducing ASCVD events in patients with established ASCVD or T2D with additional CV disease risk factors and controlled LDL cholesterol on statin therapy but who still have TGs >1.69 mmol/l.