







Diabetes is a multifaceted metabolic disorder affecting the glucose status of the human body. Impaired glucose tolerance and hyperglycaemia are the main clinical and diagnostic features and the result of an absolute or relative insulin deficiency or resistance to its action. Chronic hyperglycaemia associated with diabetes can result in end organ dysfunction and failure which can involve the retina, kidneys, nerves, heart and blood vessels.1 The clinical relationship between diabetes and atherosclerotic cardiovascular disease are well established, with the risk for cardiovascular disease (CVD) being significantly elevated in patients with diabetes.2,3
Moreover, CVD typically occurs one to two decades earlier in people with diabetes, with more aggressive, severe and diffuse distribution.4,5 The first WHO global report on diabetes published in 2016 demonstrates that the number of adults living with diabetes has almost quadrupled since 1980 to 422 million adults and this is expected to rise to 552 million by 2030.6,7 Thus, the need for effective novel therapeutic approaches for the treatment and/or prevention of diabetes and atherosclerotic disease is crucial.
Traditionally, the majority of cases of diabetes fall into two broad pathogenetic categories, type 1 (T1D) and type 2 (T2D). However, in some people this rigid classification is not applicable because other genetic, immunological or neuroendocrinological pathways are involved in its pathogenesis. T1D is related to an absolute lack of insulin due to a vaguely understood mechanism, where an immune-mediated destruction of pancreatic beta cells is the hallmark of the disorder, with hyperglycaemia only emerging when more than 90% of the beta cells are lost.8 T2D is the most common form of diabetes, accounting for 90–95% of cases. Its development is secondary to a relative insulin deficiency but the primary defect is insulin resistance.9
Various proposals and hypotheses have been developed to describe the mechanisms which are usually involved in the propagation of diabetes, mainly focusing on T2D. The increase in prevalence of the condition has been related to well-recognised risk factors, such as the adoption of a western lifestyle, sedentary lives, lack of physical activity and an energy-dense diet.10,11 Genetic predisposition, ethnicity and ageing are not modifiable risk factors for T2D, while others, such as being overweight or obese, an unhealthy diet, insufficient physical activity and smoking are modifiable through behavioural and environmental changes. However, increasing evidence has shown that inflammatory pathways are the principal, common pathogenetic mediators in the natural course of diabetes under the stimulus of the risk factors described above.12
In this article, we will highlight the emerging role of inflammation in the pathophysiology of diabetes and we will analyse the implicated inflammatory pathways and biomarkers of inflammation in diabetes and metabolic diseases. The focus of this article is to provide an overview of the current state of knowledge on anti-inflammatory therapies for diabetes, along with perspectives on future therapies for the disease.
Historical Perspectives
Observational studies provided the first evidence for the possible association between inflammation and diabetes. Over a century ago, the administration of high doses of sodium salicylate led to decreased glycosuria in people with a suspected or definite diagnosis of diabetes.13,14 Later studies on the role of inflammation in diabetes, revealed that this hypoglycaemic action was related to the inhibition of the serine kinase IkappaB kinase-beta (IKKbeta), which correlates with the post-receptor action of insulin.15
A landmark study to correlate inflammation with diabetes was conducted in animal models by Hotamisiligil et al., in 1993 and it revealed that the role of tumour necrosis factor-alpha (TNF-alpha) in obesity and particularly in insulin resistance and diabetes.16 Epidemiologic associations of inflammation with obesity and T2D were made when circulating concentrations of markers and mediators of inflammation and acute-phase reactants including fibrinogen, C-reactive protein, interleukin (IL)-6, plasminogen activator inhibitor-1, sialic acid and white cells, have been shown to be elevated in these conditions.17–21 Over the next decades, numerous studies on human and animal models provided further supporting evidence for the role of inflammation in the initiation and progression of diabetes.12,22 Accumulative evidence suggests that chronic activation of pro-inflammatory pathways in target cells of insulin action may contribute to obesity, insulin resistance and related metabolic disorders including T2D.22 The identification of potential pathways connecting inflammation to diabetes has produced growing interest in targeting inflammation to help prevent and control diabetes and related conditions, as well as improving risk stratification for diabetes by using inflammatory biomarkers as potential indexes.23,24
Inflammation in Type 1 Diabetes
T1D is an autoimmune disorder characterised by a selective, specific destruction of insulin-producing pancreatic beta cells, without apparent pathological alterations of other Langerhans cells.25 However, T1D shows significant heterogeneity in regard to the age of onset, severity of autoimmune response and efficacy of therapy, while it has also been demonstrated that both humoral and cellular immunity is involved in the pathogenesis of T1D.26–28 The first theories about predisposition support that environmental trigger factors in early life, such as infections, nutrition and chemicals that are able to activate self-targeting immune cascades, remain applicable even though the initial event is still unclear.29,30
Inflammatory Infiltrates in Type 1 Diabetes
Progress in understanding the pathophysiology of T1D has been made in parallel with the advances in the field of immunology. The predominant theory is that the beta cell pancreatic islets in patients with T1D are inflamed, called insulitis, through the course of T1D. Anderson et al., demonstrated that failure in both central and peripheral immune tolerance mechanisms contribute to the emergence of autoreactive T cells in the periphery of non-obese mice with diabetes.31 Regulatory T cells (Tregs) have been shown to also be defective in this autoimmune disease setting, along with evidence from animal models demonstrating the participation of both CD4+ and CD8+ T cells (effector T-cells/Teff) in the development of T1D as they target several beta cell autoantigens and related peptide epitopes.32–35 Moreover, by using adoptive T-cell transfer models of T1D, it has been demonstrated that T-cell subtypes are capable of inducing destructive peri-islet inflammatory infiltrate and overt diabetes.36,37 This was further depicted in human studies using pancreas samples obtained post mortem from subjects diagnosed with recent-onset T1D.27,38
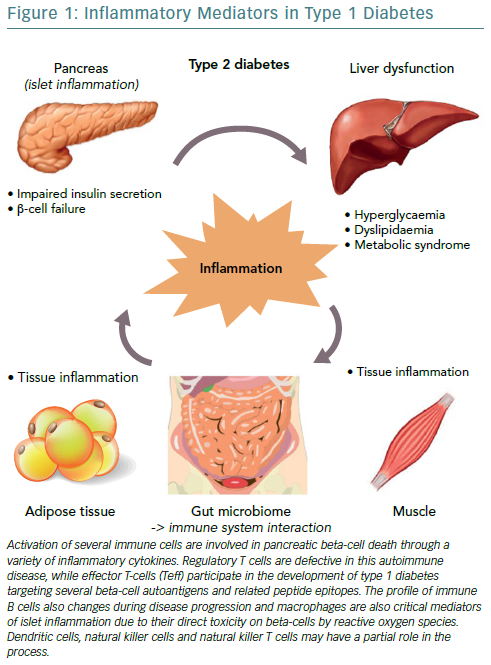
Interestingly, the immune B cell (CD20+) profile also changes during disease progression, as initial studies found they align closely with the migration of CD8+ T cells, following two different patterns, either that of high or low infiltration in islets as reported by Wilcox et al.27,38 Macrophages are also critical mediators of islet inflammation due to their ability to secrete cytokines, such as Interleukin 1 beta (IL-1beta) and tumour necrosis factor alpha (TNF-alpha) and produce reactive oxygen species (ROS).27,39 Additional studies have shown that the surrounding pancreatic exocrine tissue is abundant in both lymphocytes and neutrophils in T1D and it is suspected that these cells might also contribute to the evolution of disease.40,41 In some studies, dendritic cells, natural killer (NK) cells and NKT cells have also been found in the islet infiltrate and may have a partial role in the whole process, however, it seems that overall the interaction among different cell types regulates diabetes progression.42,43
Mediators of Inflammation in Type 1 Diabetes
The three cytokines that seem to be implicated in the inflammation of pancreatic beta cells in T1D, are the synergic action of interferon gamma (IFN-gamma) and the innate inflammatory cytokines TNF-alpha and IL-1beta.44 The combined action of these inflammatory molecules results in the upregulation of inducible nitric oxide synthase (iNOS), with subsequent production of nitric oxide (NO).45 However, even though ROS plays a role in beta cell destruction, more recent studies have demonstrated that NO is not implicated in the damage of pancreatic beta cell.46 Furthermore, studies demonstrating that the biology of the beta cell could directly influence the response to an inflammatory environment, through specific gene-guided modulation of beta cell apoptosis induced by IFN-gamma modulated by the PTPN2 gene (Figure 1).47
The mechanisms mentioned above strongly suggest that multiple pathways may exist which can contribute to pancreatic beta cell death. During this process, the control and regulation of local inflammatory cytokines production are likely to be critical factors in determining the outcome of the autoimmune progression. The disruptive effects of inflammatory and autoimmune-mediated pancreatic islet attack may lead to a vicious cycle where initial cytokine stress may urge the metabolic stress and an additional loss in beta cell function.48
Anti-Inflammatory Trials on Type 1 Diabetes
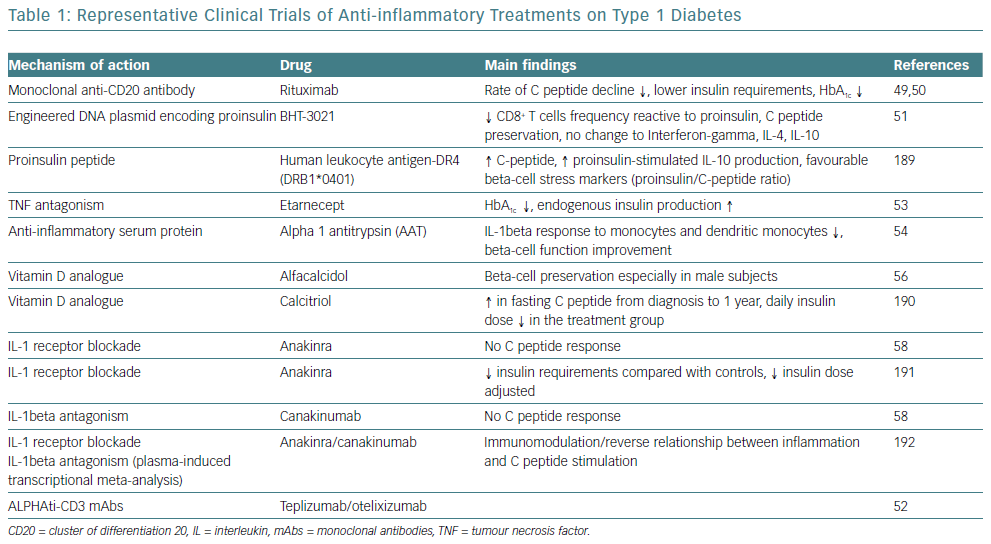
Given the obvious genetic influences in the initiation and progression of T1D, the immune cell type and the pattern that occurs in any given patient offers an important perspective on the design of clinical trials intended to slow or terminate the progression of the disease. Two initial clinical trials with rituximab, a monoclonal anti-CD20 antibody, were only partially successful.49,50 Furthermore, strategies are being developed targeting the antigen-specific T-cell response, such as the application of plasmid DNA (pDNA) vaccination with promising results.51 Moreover, two humanised anti-CD3 monoclonal antibody (mAbs), teplizumab and otelixizumab, have been evaluated in people with new and recently diagnosed T1D and showed a reduced rate of loss of beta cell function in the majority of participants.52
Cytokines are another promising target for therapy for T1D, given their involvement in the process of beta cell pathology. IL-1beta and TNF-alpha appear to be attractive initial targets for designing clinical trials based on this concept. A pilot study examined the effects of an anti-TNF-alpha therapy, etanercept, on paediatric patients newly diagnosed with T1D and demonstrated an increased endogenous insulin production and better metabolic control.53 Administration of alpha-1 antitrypsin (AAT), an anti-inflammatory serum protein, to a small group of people with T1D resulted in a reduced IL-1beta response in monocytes and dendritic cells and improved beta cell function.54 Furthermore, given the broad anti-inflammatory properties of vitamin D, it has also been identified as a potential therapeutic target.55 However, small studies of vitamin D supplementation in recent onset T1D have only resulted in modest beta cell protection.56,57
On a larger scale, interleukin-1 receptor antagonist (IL-1RN) and human monoclonal IL-1beta antibody were employed in two randomised, placebo-controlled trials in people with recent onset T1D.58 Canakinumab and anakinra were found to be safe but they were not effective as single immunomodulatory drugs in recent-onset T1D and they did not result in preserved beta cell function, as measured by stimulated C-peptide area under the curve (Table 1).
In conclusion, the immunotherapeutic trials that have been completed in human T1D have always focused on patients after clinical onset of diabetes, well after the establishment of targeted adaptive immune responses towards beta cell islets. Targeting these factors is likely to preserve remaining beta cell function, but curative treatments can only be realistically achieved by attempting at the same time to replace part of the beta cell mass that has been lost during the autoimmune process.
Metabolic Disorders and Inflammation in Type 2 Diabetes
Inflammation in Type 2 Diabetes
Several pathophysiological studies have strengthened our understanding of insulin resistance and secretion in the course of disease onset and progression.59,60 Subjects at risk of T2D display an initial state of insulin resistance compensated by hypersecretion of insulin in the beta cells. However, in the clinical course of the disease this pancreatic functional reserve is eventually unable to cope with the required insulin secretion and by the time diabetes is diagnosed, beta cells are no longer able to secrete enough insulin.61 Although the relative contribution of beta cell dysfunction and insulin resistance can vary in people with T2D, it is generally accepted that abnormal insulin sensitivity precedes the clinical diagnosis of diabetes by up to 15 years.62 Therefore, along with mechanistic studies investigating mechanisms forming the basis of insulin resistance, more recent research has also focused on the pathways leading to beta cell failure.63
The Role of Adipose Tissue and Obesity
There has been intensive research conducted into the pathophysiology of diabetes and its association with obesity and the biological role of adipose tissue. As addressed before, insulin resistance is a key component in the course of T2D. Liver and muscles have long been recognised as major contributors of systemic insulin resistance.64 Fat accumulation in the liver (steatosis) precedes overt T2D, is commonly associated with obesity and is considered a major determinant of the reduced hepatic insulin sensitivity resulting in fasting hyperglycaemia.65–67 Furthermore, it is now well accepted that the accumulation of energy due to excessive calorie intake and the lack of physical activity leads initially to fat accumulation in the subcutaneous tissue and later to other tissue compartments such as the liver, pancreas, muscles, perivascular and pericardium.67 This fat accumulation increases tissues’ insulin resistance, while pancreatic fat accumulation further determines beta cell dysfunction.64,68
Obesity and its associated conditions including metabolic syndrome, hypertension and dyslipidaemia, is positively associated with concentrations of inflammatory biomarkers, which are predictive of insulin resistance and the incidence of T2D and CVD.69–71 Obesity and metabolic syndrome specifically comprise a cluster of diseases associated with too much food and insufficient physical activity, conditions where sub-acute chronic inflammation is a common and potentially unifying mechanistic cause, accompanied by activation of at least two major inflammatory pathways, stress-activated Jun N-terminal kinases (JNK) and the transcription factor NF-kappaB.12,16,72–77 This inflammatory state via production of pro-inflammatory cytokines, is further amplified by adipokines, though a number of studies have demonstrated that adipokines stimulate additional inflammatory responses in obesity and promote obesity-induced metabolic and cardiovascular diseases.78
Animal studies have demonstrated that brown adipose tissue (BAT) has an important role in regulating energy and glucose homeostasis and is associated with peripheral insulin resistance and glucose levels.79–81 However, white adipose tissue (WAT) and mainly visceral WAT (around the trunk, upper body or abdomen) appears to be the major source of inflammatory markers in T2D, but also a target of the inflammatory process in people with diabetes. It produces cytokines and several other bioactive substances involved in the inflammatory pathways, such as TNF-alpha, IL-1, IL-6, IL-10, leptin, adiponectin, monocyte chemoattractant protein, angiotensinogen, resistin, chemokines, serum amyloid protein, and many others collectively referred to as adipokines.82–85 Further infiltration of adipose tissue by macrophages and immune cells (B cells and T cells) trigger local and systemic chronic low-grade inflammation, by producing more cytokines and chemokines that serve as a pathologic link between obesity, insulin resistance and diabetes.86
The Role of Gut Microbiota in Type 2 Diabetes
The role of the gut in the pathophysiology of diabetes can be approached from two different viewpoints. Studies have suggested that several mechanisms may be involved in weight loss and diabetes control after bariatric surgery, beyond malabsorption or anatomical restriction.87 Indeed, complex changes in multiple gut hormones have been shown after bariatric procedures and have been proposed as adjunctive mechanisms for short- and long-term positive metabolic effects, serving as possible novel therapeutic approaches to obesity and insulin resistance.88,89
In the past few years, a two-way relationship between the gut microbiome in the host’s energy balance and immune function has been demonstrated.90 The gut microbiome seems to differ between obese and lean subjects, flora composition influences metabolism and inversely, diet and metabolic status influence the composition of the gut flora, while a faecal microbiome transplantation from lean donors to insulin-resistant subjects results in beneficial metabolic effects.91–94 It has been postulated that products from the gut microbiome may interact with the immune system inducing a tissue metabolic modification, which feeds the molecular origin of the low-grade inflammation that characterises the onset of obesity and diabetes.95
An altered gut microbiota can directly affect immune cells in the gut and indirectly affect immune cells via microbial products including LPS, metabolites and short chain fatty acid (SCFAs), all of which can affect adipogenesis and/or insulin resistance.96–101 Lipopolysaccharide (LPS) is believed to cause low-grade inflammation mediated by the induction of inflammatory cytokines by immune cells and adipocytes, while SCFAs can modulate gene expression of human monocytes and reduce pro-inflammatory cytokine and chemokine production.102 SCFAs can also promote regulatory T-cell generation through several pathways, thereby suppressing the function of inflammatory T cells. These are able to block IFN-gamma inducible protein 10 (IP-10) release in human colonic sub-epithelial myofibroblasts, acting not only on immune cells systemically but also on intestinal tissue cells locally.103,104
The Role of Pancreatic Beta Cell Failure in Type 2 Diabetes
Independent of the aetiopathogenetic mechanism among the different types of diabetes, the common pathway seems to be the inflammation in the pancreatic Langerhans beta cell islets (insulitis), in the concept of an auto-inflammatory process, which results in reduction in both beta cell number and function.105 It has been suggested that in people with a genetic predisposition, the ‘stressed’ beta cell may stimulate local inflammation and modify the balance between beta cell mass and function in the islets of Langerhans.106,107 Several experimental models as well as observational studies in humans have demonstrated that macrophages play a key role in the islet inflammation seen in T2D.108–111 Inflammasome/IL-1beta signalling is the most common, well-studied and high-impact pathway activated in islets of multiple T2D models that cause beta cell dysfunction.112,113 It is likely that other immune cell types are involved in islet inflammation in T2D, while islet autoimmunity has also been suggested to contributes to beta cell functional decline during the course of T2D.114,115
Among factors that stimulate islet macrophages to secrete IL-1beta in vivo in human islets are amyloid polypeptide, free fatty acids (FFAs) and endocannabinoids.110,111,116 However, it has been assumed that hyperglycaemia is produced initially in the inflammation in pancreatic beta cells by inducing apoptotic mechanisms.117 ALPHA particular pathway was proposed by Maedler et al. who showed that hyperglycaemia may induce the production of IL-1beta by stimulating pro-apoptotic receptor FFAs on beta cells.118
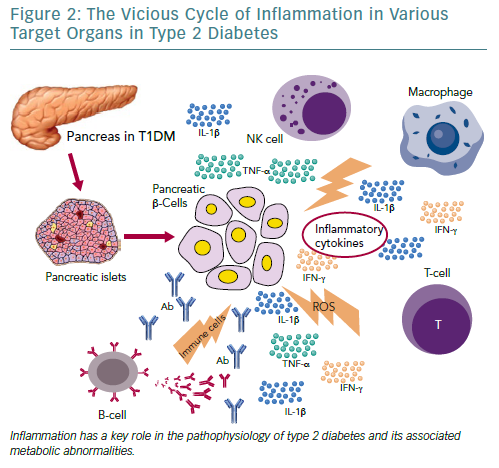
FFAs can also produce and secrete IL-1beta and IL-1-dependent pro-inflammatory cytokines in pancreatic islets and thus to reduce the inflammation. In addition, after its initial secretion, IL-1beta regulates its production in pancreatic beta cells by auto stimulation, while this process also increases nitric oxide production leading to reduction in ATP concentration in the mitochondria, which can cause further beta cell dysfunction and reduced insulin secretion.119–122 Oxidative stress may also potentiate the generation of ROS along with other pro-inflammatory cytokines and chemokines in the beta cells that disrupts the blood flow into them and destroys their function.108,123,124
Experimental studies have confirmed that IL-6 induces apoptosis in pancreatic islets together with other inflammatory cytokines and acts as a predictor and pathogenic marker for the progression of T2D.69,124,125 TNF-alpha is also considered to play an essential role, by creating a linkage among insulin resistance, obesity and islet inflammation.125 Its overproduction in adipose tissue seems to feed the inflammation and beta cell death in pancreatic islets and produces additional insulin resistance in peripheral tissues.126,127
Evidence of Inflammation in Other Organs in People with Type 2 Diabetes
Immune system activation is highly related to T2D incidence and progression and adaptive and innate immunity are involved in adipose tissue inflammation. The phenotype switching of macrophages from predominantly anti-inflammatory M2-type to increased proportions of pro-inflammatory M1-type macrophages plays a crucial role in the initiation and amplification of islet inflammation.128 However, the evidence shows that the recruitment of B cells and T cells precedes adipose tissue infiltration by macrophages.86
Moreover, several other organs have been reported to participate in the metabolic homeostasis and inflammatory state in T2D, such as the liver, the neural system and possibly skeletal muscle.129–133 However, more research is needed to support this evidence (Figure 2).
Current Knowledge on Diabetes Treatments
Drugs with Pleiotropic Effects
The current therapeutic approaches to T2D have anti-inflammatory properties in addition to their major modes of action. Non-pharmacological therapies, such as lifestyle interventions, but also pharmacological and bariatric surgical approaches for weight loss, appears to reduce inflammation assessed as circulating CRP and IL-6 concentrations, and improves cardiovascular and all-cause mortality.134–138
Statins also have anti-inflammatory properties beyond their ability to lower levels of low-density lipoproteins (LDL) cholesterol. The Justification for the Use of Statin in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) demonstrated that rosuvastatin reduced high-sensitivity CRP along with LDL cholesterol, however the effects of statins on glycaemic control are conflicting, implying that targeting inflammation with statins does not improve glycaemia and therefore does not provide an integrated anti-inflammatory approach for diabetes and CVD.139–141
Anti-diabetic agents, including insulin, have intrinsic anti-inflammatory effects associated with their primary mechanisms of action and are also associated with reductions in inflammatory markers. Insulin itself decreases NF-kappaB activity in peripheral blood mononuclear cells which reduces inflammation.142 The anti-inflammatory actions of thiazolidinediones through binding and activation to the peroxisome proliferator-activated receptor gamma (PPARgamma), seems to be related to trans-repression of NF-kappaB and reduced expression of NF-kappaB targets.143
In addition to its metabolic effects, metformin has anti-inflammatory actions that appear to be independent of glycaemia and are most prominent in immune cells and vascular tissues.144–150 Dipeptidyl peptidase-4 inhibitors (DPP-4) and GLP-1 receptor agonists also have intrinsic anti-inflammatory properties, however, beyond their anti-diabetic effects, the contribution of inflammation reduction to diabetes and cardiovascular improvements remains unknown.151–153 Finally, a new class of anti-diabetic drugs, sodium–glucose cotransporter-2 inhibitors (SGLT2 inhibitors) acts by increasing renal excretion of glucose. Preliminary data in humans demonstrate a possible improvement on the circulating biomarkers of inflammation by SGLT2-inhibitors; however, more studies are needed.152
Anti-Inflammatory Drugs in Type 2 Diabetes
Multiple medical approaches that directly target inflammatory pathways have been studied in the past few years supporting the concept of anti-inflammatory treatment for cardiometabolic diseases, such as diabetes and atherosclerotic CVD.154–156 For a long time, salicylates, especially aspirin, have been used to treat thrombosis in primary and secondary CVD prevention, as well as to treat rheumatoid diseases.157,158 They were the first class of drugs reported to lower glucose in diabetes more than a century ago, however, several studies with salicylate products have demonstrated an improved metabolic profile in patients with obesity and diabetes, suggesting a potential efficacy for diabetes prevention and control.159–166
Methotrexate is a disease-modifying drug broadly used to treat rheumatic diseases among other conditions, while its efficacy on glycaemic control was demonstrated in a small cohort study.167 The preliminary data drove the design and conduction of a large clinical trial with methotrexate among patients with previous MI and either T2D or metabolic syndrome, however, methotrexate had neutral findings on IL-1b, IL-6 and CRP levels, while more data are anticipated for the effects on T2D.168
Biological Agents as Anti-Inflammatory Therapy in Type 2 Diabetes
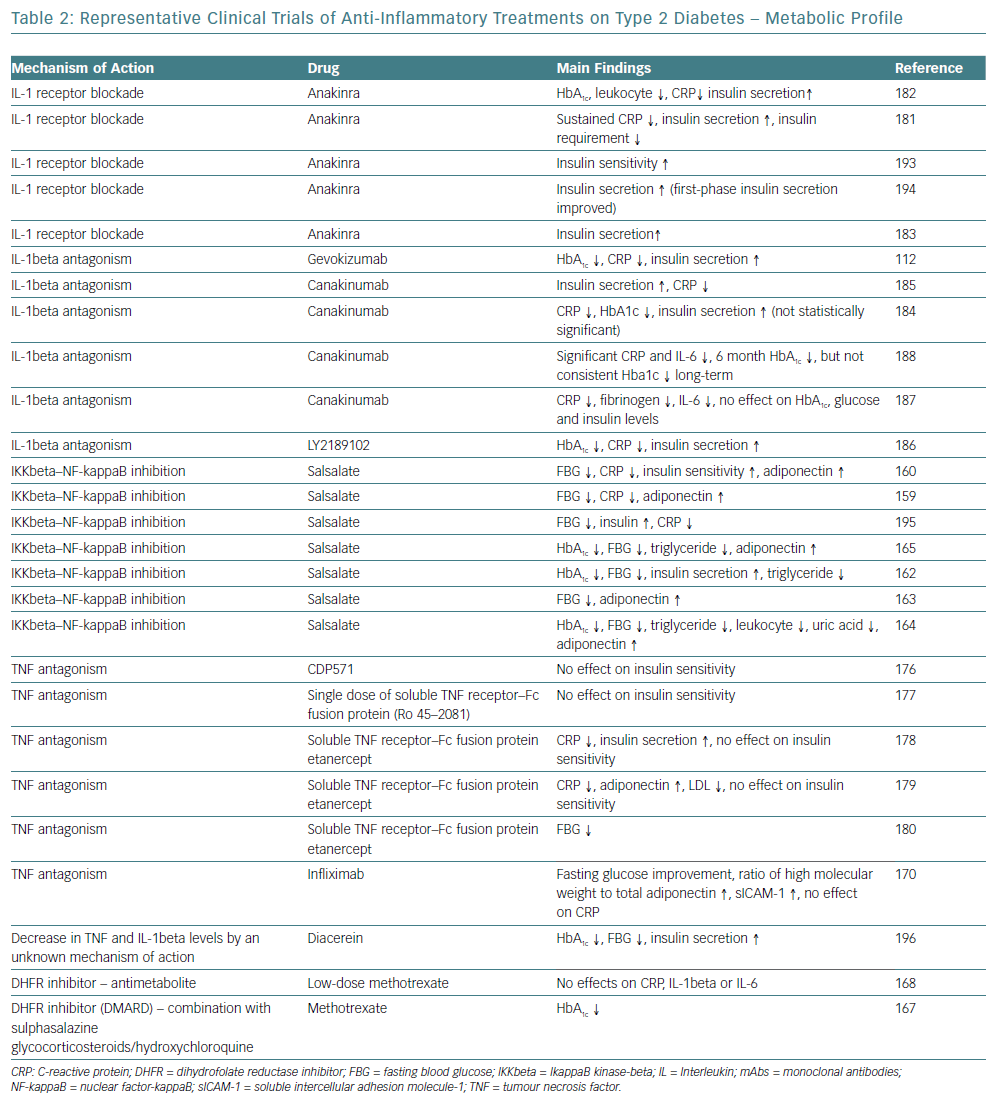
Targeting cytokine production and secretion to prevent further activation of inflammation have been proposed with the intention of stopping the initiation and progression of T2D. TNF-alpha antagonists have been used to treat inflammatory conditions and have been associated with improved glycaemic control and decreased incident of diabetes, while more studies on patients with unfavourable cardiometabolic profile did not demonstrate adequate results, with the exception of a randomised 6-month trial.169–180
The mechanism of action of IL-1beta is consistent with the pathogenesis and progression of T2D is. Improved beta cell secretory function and glycaemia, as well as reduced inflammatory biomarkers in people with diabetes and pre-diabetes have been demonstrated by IL-1beta antagonists, such as anakinra and gevokizumab.112,120,181–6 Studies on CVD and atherosclerosis prevention with IL-1beta antagonists have also been conducted. One study showed that canakinumab reduced the inflammatory proteins CRP, IL-6, and fibrinogen in persons with T2D and high cardiovascular risk with no effect on HbA1C, glucose, and insulin at 4 months, while the large randomised trial with canakinumab – Canakinumab Anti-Inflammatory Thrombosis Outcome Study (CANTOS) – over a median period of 3.7 years did not reduce the incidence of diabetes in patients with prior MI and high-sensitivity CRP (hsCRP) ≥2 mg/l (Table 2).187,188
Future Perspectives for the Treatment of Diabetes
Novel approaches on T2D to evaluate anti-inflammatory diets and modulate an individual’s microbiome are under study. Clinical trials investigating the effects of vitamin D supplementation on serum levels of inflammatory markers have provided inconsistent results, with no evidence of effects in most trials, or effects on selected markers in others. There are also studies investigating whether antagonists of leukotriene production enzymes – 5-lipoxygenase (5-LO), 5-LO-activating protein and LTA4 hydrolase – or receptor binding BLT1 have cardiometabolic outcome benefits, however these results have not yet been reported. The potential for targeting cholinergic pathways, immune modulation or other mediators of inflammation such as JNK and toll-like receptors (TLRs) are also being researched.
Conclusion
The increasing prevalence of diabetes makes it imperative that research should focus on its prevention as well as its treatment. An improved understanding of the mechanisms linking inflammation to diabetes and related complications has stimulated interest in targeting inflammatory pathways as part of the strategy to prevent or control diabetes and its complications.
T1D is considered to be more of an immunological response rather than a metabolic disorder and the preliminary results of trials using anti-inflammatory and immunomodulatory medication are promising. These treatments in combination with possible use of stem cells to regenerate pancreatic beta cells could potentially be the key to permanent treatment of T1D. Therefore, after a holistic review of the possible mechanisms that lead to T1D and T2D and the numerous already described inflammation pathways that are involved, it becomes more and more clear that future research should focus on simultaneous suppression of various inflammatory response pathways rather than focusing on one pathway at a time.