







Takotsubo cardiomyopathy (TTC) has gained more recognition since its first description in 1990 by Satoh et al.1,2 The word takotsubo refers to the Japanese octopus trapping pot with a large round base and narrow neck, which is the characteristic shape of the left ventricle (LV) in this syndrome, although other morphologies have since been described.3 TTC is known by many names including stress cardiomyopathy, broken heart syndrome, apical ballooning syndrome and ampulla cardiomyopathy. It is a reversible form of cardiomyopathy that presents clinically as an acute myocardial infarction (MI) triggered by an emotionally or physically stressful event. An abnormal response to a catecholamine surge leads to TTC and a stressor can be identified in a majority of cases. TTC occurs at a significantly higher frequency in post-menopausal women with 80–85 % of all cases presenting in women. Less than 3 % of cases occur in those less than 50 years of age.4
The exact incidence of TTC is unknown since it is likely under-diagnosed. The annual rate is between 7,000–14,000 cases per year in the US and it is estimated that 1–2 % of acute coronary syndrome (ACS) diagnosed in the US is actually TTC.4,5 The Nationwide Inpatient Sample (NIS) database in 2008 found that 0.02 % of all hospitalisations in the US were attributed to TTC.5 This study also found that hyperlipidaemia, active smoking, alcohol abuse, anxiety and stress were associated with TTC.5 One study found significantly lower cardiovascular risk factors in TTC patients compared with population-matched MI patients.6 On the contrary, the recently published systematic review of TTC patients, known as the COmorbidity freqUency iN Takotsubo Syndrome (COUNTS) study, reported a high prevalence of cardiovascular risk factors in TTC patients compared with the general population.7,8 Out of the 1,109 patients in this study, on average 17 % were obese, 54 % had hypertension, 32 % hyperlipidaemia, 17 % were people with diabetes and 22 % were smokers.7 Common comorbidities reported in TTC patients include malignancy, neurological disorders including stroke, pulmonary diseases, chronic kidney disease, thyroid disease and psychological disorders.7 Unlike the study by El-Sayed et al., the COUNTS study reports a poor association between TTC and drug abuse, chronic liver disease and sepsis.6,7 Interestingly, TTC cases occurred at a higher rate in July in contrast to acute MIs, which peak in occurrence in winter months.5 The exact reasons for this potential seasonal variation in TTC are unknown.
The Mayo Clinic proposed criteria in 2004 for diagnosing TTC (see Table 1).9 They suggested that all four criteria should be present in order to diagnose TTC. In 2006 the American Heart Association (AHA) classified TTC as a primary acquired cardiomyopathy.
Precipitating Factors
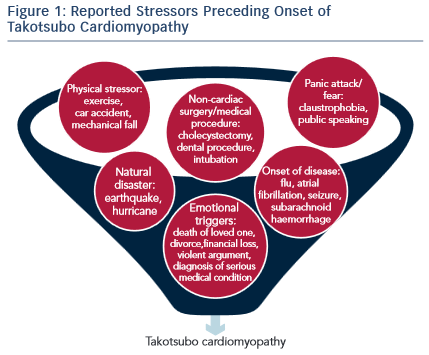
In most cases, TTC presents shortly after the individual experiences a major emotional or physical stressor (see Figure 1). A prospective study of 136 patients presenting with TTC found that 47 % of the patients experience an emotional stressor, 42 % a physical stressor and 11 % had no evidence of any stressor.10 In the COUNTS study, 39 % patients experienced a preceding emotional trigger and 34 % patients a physical stressor.7 Emotional stressors range from public speaking, work conflicts/job concerns, anger, to hearing bad news such as death of a family member or a pet. Physical stressors include medical illness such as undergoing surgery or chemotherapy, a fall or drug use. Even though a clear precipitating stressor could not be identified in some TTC patients, the lack of a stressor does not exclude the possibility of diagnosing TTC.
Certain drugs are shown to be precipitating factors in TTC. Out of 58 patients with case reports of drug-induced TTC, 45 were due to medications that cause direct or indirect stimulation of the sympathetic nervous system (SNS).11 These included agents such as dobutamine, epinephrine, norepinephrine, ephedrine, ergonovine, atropine, nortriptyline and oxymetazoline.11 Levothyroxine has been linked to TTC, and although it does not act on the SNS, the cardiac effects of levothyroxine mimic those caused by catecholamines. Pazopanib is a vascular endothelial growth factor receptor antagonist that inhibits nitric oxide production, and increases sensitivity to catecholamines.11 Agents with no clear link to catecholamine activity including dypyridamole, potassium chloride, lumiracoxib, 5-fluoruracil and combretastatin have also been implicated in TTC.11
Mechanisms of Catecholamine Toxicity
It is evident that an abnormal response to a catecholamine surge leads to the development of TTC, but the mechanisms by which it does so have not been clearly established (see Figure 2). Introducing rats to a stressful situation such as immobilisation induces the typical apical ballooning pattern of TTC.12 It has been shown that pre-treatment with a beta-blocker, alpha-blocker or a combination of the two prevented the occurrence of TTC in immobilised rats.12 Furthermore, giving rats an alpha or beta agonist actually induced TTC.13 Plasma catecholamine levels in patients that have undergone a major emotional or physical stressor with subsequent TTC are found to be at supraphysiological levels 1–2 days after the initial stressor and half the peak values after 1 week.14 These levels are twice as high as are observed in patients with Killip class III MI.14 There is evidence of increased sympathetic activity in patients with TTC as confirmed by 123I-meta-iodobenzylguanidine (123I-mIBG) single-photon emission computed tomography (SPECT) imaging.15 Additionally, the plasma levels of catecholamine precursors and neuronal as well as extraneuronal breakdown products are elevated, implicating increased catecholamine synthesis as well as neuronal and extraneuronal metabolism.14
Epicardial coronary artery spasm causing ischaemia has also been implicated in TTC. The elevation of catecholamine levels stimulates the α1 receptors on the coronary vasculature leading to coronary vasoconstriction with resultant ischaemia. This is supported by ST elevations being a common finding in TTC despite the absence of obstructive coronary artery disease. However, the ST elevations in TTC are more diffuse and do not typically follow a distinct epicardial distribution. Thus, it is proposed that multi-vessel coronary vasospasm may be responsible for the development of TTC. Tsuchihashi et al. demonstrated that 21 % (n=48) of patients have inducible vasospasm with use of acetylcholine.16 Another systematic review showed 27.6 % of subjects have provoked coronary vasospasm in response to acetylcholine or ergovine.17 Thus, while an important contributor towards the development of TTC, multi-vessel epicardial coronary spasm does not explain the majority of TTC cases.

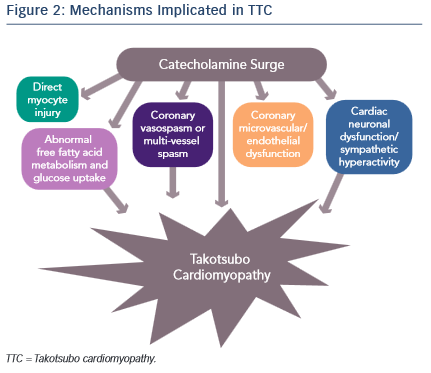
Alternatively, coronary microvascular and endothelial dysfunction is another hypothesised mechanism of TTC. One of the major arguments for this mechanism is that electrocardiogram (ECG) changes are diffuse and not suggestive of a single coronary artery territory and on cardiac catheterisation the majority of patients do not have obstructive coronary artery disease. Additionally, the peak troponin level and the extent of ECG changes in patients with TTC are shown to correlate to the severity of endothelial dysfunction.18 These patients are found to have a reduced coronary blood flow during the acute TTC event that remains impaired after the resolution of TTC left ventricular dysfunction, suggesting that coronary microvascular dysfunction is present at baseline in patients with TTC.19,20 Moreover, female patients with a history of TTC have evidence of impaired microvascular dilation in response to acetylcholine as well as a significantly lower increase in peak coronary blood flow compared with age-matched women with normal microvascular responses.20
The reactive hyperaemia peripheral arterial tonometry (PAT) is a reflection of changes in peripheral endothelial function and correlates with coronary endothelial function. TTC patients compared with post-MI and post-menopausal control women demonstrate more adverse mental stress PAT scores compared with the other groups, consistent with a persistently abnormal physiological response in TTC patients.21 Excessive vasoconstriction and impaired endothelium dependent vasodilation is suggested to cause this abnormal response to mental stress testing.20
It has previously been proposed that left ventricular outflow tract (LVOT) obstruction leads to TTC. The theory is that catecholamine release causes LOVT obstruction thus increasing the mechanical stress on the cardiac apex and subsequently leads to myocardial stunning.22 A recent study reveals that dobutamine causes mid-ventricular outflow gradients in both patients with and without a history of TTC.22 This means that there are no differences in patients with or without a TTC history that would predispose them to develop TTC. It is not likely that LVOT obstruction is the sole cause of TTC given that the incidence of LVOT obstruction in TTC is about 20–25 %.23
A fourth proposed mechanism is that the elevated catecholamine levels may cause direct myocyte injury. In previous studies it is shown that norepinephrine causes an increase in cyclic adenosine monophosphate (cAMP)-mediated intracellular calcium overload that is responsible for myocyte toxicity.24 Furthermore, catecholamines can be a source of oxygen-derived free radicals that then interfere with intracellular calcium transporters, leading to excess calcium influx and subsequent myocyte damage.14,25 One way in which increased intracellular calcium may cause cell injury is through the high-energy phosphate deficiency that results from excessive activation of the calcium dependent ATPase, therefore leading to impaired mitochondrial function.24 Histologically, myocytes have been shown to have contraction band necrosis and mononuclear inflammation, both characteristic signs of catecholamine toxicity that differ from ischaemic-induced necrosis.25 Contraction band necrosis and mononuclear inflammation are also seen in other catecholamine-excess states, such as subarachnoid haemorrhage and pheochromocytoma.25 It has been hypothesised that the fibrosis seen in TTC does not reach a critical value due to the concomitant release of anti-fibrotic factors by norepinephrine, and is the reason that TTC is reversible.25
Another theory of acute-myocardial stunning via alterations in cell signalling is proposed as a further cause of the direct effect of catecholamines on the heart. Normally, norepinephrine binds to the β1-adrenoreceptors that are coupled to Gs proteins, subsequently increasing cAMP to activate protein kinase A and therefore cause increased contractile response via release of calcium. Epinephrine has the same effect but with a higher affinity for the β2-adrenoreceptors, which also leads to a positive inotropic response. At supraphysiological epinephrine levels, the β2-adrenoreceptors couple to Gi protein instead of the Gs protein therefore leading to a negative inotropic effect on the heart.2 Additionally, although there are more sympathetic nerve endings in the basal myocardium, there is a higher concentration of β-adrenoreceptors at the apex thus explaining the typical apical ballooning pattern due to apical akinesis.26 Once the epinephrine levels return to normal, the β2-adrenoreceptors are once again coupled to the Gs protein.2 The switch to Gi protein may have a protective role in the myocardium. High levels of β1-adrenoreceptor-mediated Gs activation can stimulate apoptotic pathways therefore switching to Gi protein via β2-adrenoreceptors inhibits apoptosis from occurring.27 Additionally, as previously mentioned, intracellular calcium overload by the Gs receptor can be cardiotoxic, so the switch to the Gi pathway may be cardioprotective.
Neurogenic stunned myocardium is another mechanism proposed to cause TTC. The fact that an emotional stressor causes TTC suggests that the brain plays a role in inducing cardiac injury. Patients with subarachnoid haemorrhage are found to have elevated catecholamine levels. These patients, particularly females, demonstrate similar findings as those in TTC such as diffuse ST elevation without obstructive coronary artery disease on cardiac catheterisation, significantly reduced wall motion of the apex, and histological changes consistent with those of catecholamine excess, such as contraction band necrosis.28,29 In subarachnoid haemorrhage, ischaemia of the hypothalamus releases excess amounts of norepinephrine and subsequently leads to myocyte stunning.28 A more recent study with three patients shows direct evidence that brain activation is implicated in TTC by measuring cerebral blood flow via SPECT.30 Cerebral blood flow was increased in the hippocampus, brainstem and basal ganglia and decreased in the pre-frontal cortex during the acute phase of TTC in all three subjects.30 This pattern of brain activation is is found in acute stress and with sympathetic activation.30
In a recent study by Vaccaro et al., TTC patients are found to have increased SNS activity at baseline and a decreased baroreceptor response compared with congestive heart failure (CHF) patient controls.31 CHF patients have elevated SNS activity, thus this study demonstrated that TTC patients have even higher sympathetic activity. Moreover, TTC patients also had decreased baroreceptor inhibition of the SNS. In this study, SNS activity was directly measured by assessing muscle sympathetic nerve activity (MSNA).31 The study only determined an association between increased SNS and decreased baroreceptor response but did not determine causality. Also, by looking at the musculature, the study focused on the SNS activation of the peripheral vasculature but not the heart itself. However, it does suggest that autonomic dysfunction could play a role in TTC. This can further be supported by the fact that many patients with haemorrhage or ischaemic stroke have high occurrence of TTC, and may serve as an alternative explanation for neurogenic stunning.
Clinical Presentation
The clinical presentation of TTC is indistinguishable from acute MI in terms of symptoms, elevation of cardiac enzymes and ECG changes. The most common symptoms upon presentation are anginal chest pain and dyspnea. Syncope can also be a presenting symptom but is less frequently observed. Patients can less commonly present with arrhythmias, sudden cardiac death, asystole or cardiogenic shock. There are many ECG patterns described in TTC, most prominently ST elevations and/or diffuse T wave inversions, and prolongation of the QTc interval.3,4,17 The ST elevations occur primarily in the precordial leads therefore mimicking an acute anterior MI.4 The ST elevations typically resolve after 2–3 days and rarely will patients develop pathological Q waves. Similarly, the T wave inversions are also transient and resolve within 3–4 months. Other less-frequent ECG findings include ST depressions, new-onset bundle branch block, high-degree atrioventricular (AV) block, or simply a normal ECG.3 Additionally, troponin is often elevated, which further makes TTC difficult to distinguish from ACS. Multiple studies have noted that the extent of troponin T elevation is less than that of STEMI and inexplicably low for the wall motion abnormalities seen with TTC.4 Furthermore, the magnitude of troponin elevation does not correlate with the ECG pattern, clinical features, LV contraction pattern or patient outcomes.10
Imaging
For the typical presentation of TTC, imaging of the LV shows apical and mid-wall hypokinesis with a hypercontractile basal myocardium.2,3 Atypical patterns of LV wall motion abnormalities exist in TTC and are increasingly recognised, which include isolated basal ballooning, mid-ventricular ballooning with basal and apical sparing and biventricular ballooning.3 The most common atypical form is the ‘inverted Takotsubo’ pattern that consists of basal ventricular akinesis with normal apical function.2,3,17
Cardiac magnetic resonance imaging (CMR) has been gaining popularity as a diagnostic modality for TTC. In a large prospective study consisting of 256 TTC patients, 81 % were found to have myocardial oedema correlating to areas of wall motion abnormalities.3 Myocardial oedema in TTC is global, and the amount of myocardial oedema in a particular area predicted the extent of wall motion abnormality.32 Although decreased, myocardial oedema was still present after 3-month follow-up. The extent of myocardial oedema correlated with that of peak N-terminal of the prohormone brain natriuretic peptide (NT-proBNP) levels, a marker of inflammation and NT-proBNP values also remained elevated for 3 months.32 This suggests inflammation as a cause of the oedema, and that the inflammatory response persists even after recovery of systolic function.32 Late gadolinium enhancement (LGE), indicative of myocardial scarring, is present in 9 % of TTC patients. Eitel et al. have created the following diagnostic criteria for diagnosing TTC via CMR: severe left ventricular dysfunction in a non-coronary distribution pattern, myocardial oedema collocated with the regional wall motion abnormalities, absence of high signal areas in LGE images and increased early myocardial gadolinium uptake.3
Treatment
There are no specific guidelines for the treatment of LV dysfunction in TTC. Physicians generally follow the AHA/American College of Cardiology (ACC) treatment guidelines for ACS/non-ST segment elevation myocardial infarction (NSTEM/STEMI), as the syndrome’s presentation mimics ACS.33,34 Although only found in 20 % of patients, left heart failure is the most common complication of TTC.4 Angiotensin-converting-enzyme (ACE) inhibitors and diuretics are used for LV dysfunction and treatment of acute heart failure as needed.35 Patients are also started on an alpha and beta-blocker given the catecholamine surge in TTC. However, some authors do not recommend beta-blockers due to the concern of unopposed alpha stimulation in the setting of catecholamine excess.36
In the setting of cardiogenic shock, a differentiation should be made between pump failure and LVOT obstruction. If LVOT obstruction is present, then preload and afterload optimisation with the use of beta- blockers is recommended. Propranolol was shown to significantly increase left ventricular ejection fraction (LVEF) in patients with LVOT obstruction, but not in those without it.23 Beta-blockers will reduce the ventricular gradient and therefore reduce hypotension and increase LVEF. If dobutamine is given in the setting of LVOT obstruction, it will worsen the ventricular gradient and subsequently exacerbate hypotension.23 In cases of cardiogenic shock from primary pump failure, inotropic therapy did not show any benefit due to stimulation of the beta-2 and alpha adrenergic receptors, which can further exacerbate TTC.37 As a result, there should be early consideration of the possibility of intra-aortic balloon pump use. In some case reports, the use of levosimendan, a non-catecholamine inotrope, is shown to be beneficial in treating patients with cardiogenic shock due to pump failure.38,39 However, no large-scale studies have been carried out to date to prove efficacy.
Given apical hypokinesis in TTC, echocardiography and often cardiac MRI are needed to evaluate for LV thrombus. Anticoagulation is considered in the initial stages where there is severe LV dysfunction, and especially if thrombus is identified in the LV apex.4 Anticoagulation should be used with caution due to the increased risk of LV rupture with apical ballooning.36 There are no current guidelines on the duration of therapy with anticoagulation. Patients with recurrent episodes can be continued on beta-blocker therapy in addition to stress management.
Natural History and Prognosis
While TTC has a good prognosis, it is by no means a benign disease. In the majority of cases, the LV wall motion abnormalities resolve in days to weeks. Systolic function can take longer to normalise and on average resolves over 4–10 weeks.4,10,40 One study that followed patients with TTC demonstrates that the rate of recurrence is 2.9 % per year within the first 4 years, and 1.3 % per year subsequently.40
In-hospital mortality from TTC in the acute phase is generally around 1 % though it can be as high as 9 %.4,10,41,42 Independent predictors for mortality in TTC include the presence of certain comorbid conditions and male gender.41 A study by Brinjikji et al. using the NIS from 2008 to 2009 demonstrated that in-hospital mortality was significantly higher in males than in females: 8.4 versus 3.6 % respectively.41 Males also had a higher incidence of developing life-threatening complications including cardiogenic shock and cardiac arrest as well as dying from these complications, whereas females were more likely to develop acute CHF. However, out of the patients who experienced in-hospital mortality, over 80 % consisted of those with concomitant critical illnesses, such as sepsis, acute renal failure, stroke and respiratiory insufficiency.41 Since men had more underlying critical illnesses than women, this could explain their higher mortality rate and higher rates of developing and dying from acute complications.41 Furthermore, significantly more males present initially with cardiogenic shock or out-of-hospital cardiac arrest.43
Cardiogenic shock occurs in 4–20 % of patients with TTC,41,44 as a result of ventricular failure or LVOT obstruction. If LVOT obstruction is responsible for cardiogenic shock, it is usually accompanied by systolic anterior motion (SAM) of the mitral valve. Mortality from cardiogenic shock with TTC is reported to be as high as 16 % in some cases.41 Ventricular arrhythmias occur in 4–9 % of TTC patients in the acute phase. Of those that develop ventricular fibrillation or cardiac arrest, mortality is reported to range from 1 % to as high as 27 %.41
Interestingly, causes of long-term mortality differ in patients with a history of TTC compared with patients with ACS: a majority of TTC patients die of non-cardiac causes in the long term. Sharkey et al. reported that during an average follow-up period of 1.3 years, 17 out of the 133 TTC survivors died, with none being from a cardiac cause.10 In another study by Song et al., with a mean follow up of 42 months, mortality from non-cardiac causes was significantly higher than that from cardiac causes: 21 versus 2 %, respectively.42 Non-cardiac diseases, such as malignancy and stroke, were found to be an independent risk factor for long-term mortality.42 This is different from ACS patients in whom long-term mortality is usually from cardiac causes.45
Increasing evidence for an association between malignancy and TTC exists. One study shows TTC patients to have a significantly higher incidence of any malignancy compared with population-matched MI and orthopaedic patients.6 Another study demonstrated that 18 % of TTC patients versus 3 % of MI patients have a history of malignancy at the time of the event.45 Subsequently, seven patients in the TTC group versus zero patients in the MI group went on to develop cancer at the follow-up time of about 1.6 years.45 Additionally, in the COUNTS study, 10 % of patients have a malignancy.7 The association of malignancy with TTC brings up the possibility that TTC may be a manifestation of a paraneoplastic syndrome,7,43,45 which may imply that TTC is neither as benign as previously believed, nor does it have as favourable a long-term prognosis.
Why Predominantly Women?
TTC affects a disproportionately greater number of post-menopausal women compared with pre-menopausal women and age-matched men.5 There are several mechanisms proposed to explain this sex difference. It has been reported that catecholamine stress induces upregulation of immediate early genes (IEGs), certain proto-oncogenes and heat shock proteins that are not shown to be activated during reperfusion after an ischaemic episode.13 Oestrogen minimises the catecholamine-induced upregulation of IEGs, genes that are transiently activated to rapidly adapt to a stressor.13 Supplementation with oestradiol decreased the stress-induced upregulation of IEGs in rat models. Oestradiol-supplemented ovariectomised rats did not show any significant reduction in LV contraction whereas ovariectomised rats without supplementation with oestradiol showed a significant reduction in LV contraction.46 Therefore, oestrogen counteracts the cardiac effects of the SNS by decreasing the production of IEGs.8
Oestrogen has also been implicated in maintaining appropriate glucose uptake for cardiac energy. A recent report indicates that the female heart depends on glucose as its energy source more than the male heart.47 The female heart most efficiently utilises glucose between ages 51–70 and after age 70, glucose uptake is reduced.47 The relative lack of oestrogen as women age, therefore, may potentiate this attenuated glucose uptake thus predisposing post- menopausal females to TTC. Males are not predisposed to TTC despite their relative lack of oestrogen because they are not as dependent on glucose as their preferential cardiac energy substrate.47
Oestrogen may also play a role in enhancing the β-adrenoreceptor sensitivity and in promoting vasodilation. Post-menopausal women have a decreased β-adrenoreceptor responsiveness to catecholamine stimulation than younger females.48 However, their α-adrenoreceptor vasoconstriction response to catecholamines remains the same.48 As a result, there is more β-adrenoreceptor stimulation in relation to β-adrenoreceptor responsiveness thus leading to more vasoconstriction, which in the setting of endothelial dysfunction may trigger TTC. Additionally, oestrogen indirectly increases the production of nitric oxide thus promoting vasodilation.8 This nitric oxide-induced vasodilation helps minimise the effect of catehcholamines, especially in the microvasculature.8
Stollberger et al.49 discuss two opposing speculations related to the sex difference in TTC: 1) males are better protected biologically against stress and 2) males are biologically less resistant than females against stress. Historically, men were exposed to more physical stressors than women and therefore may be better protected biologically than women.49 Additionally, males have a higher density of adrenergic receptors compared with women and can thus protect themselves from catecholamine excess better than females.5 On the other hand, males have a higher rate of sudden cardiac death and therefore possibly die more frequently from the acute LV dysfunction and thus die before they are diagnosed with TTC.49
Conclusion
TTC is an acute, reversible form of catecholamine stress-induced cardiomyopathy that mimics acute MI, and predominates in post- menopausal women. It presents with signs and symptoms of ischaemia and acute left ventricular dysfunction with regional wall motion abnormalities in the setting of no obstructive coronary artery disease. An emotional or physical stressor usually precedes TTC. The syndrome has a good prognosis although a few percentage of patients experience recurrent events. Mechanisms implicated in TTC include multi-vessel coronary spasm, endothelial and coronary microvascular dysfunction and direct catecholamine toxicity. Clinicians should be aware of this syndrome and studies that investigate mechanistic pathways of TTC may help with development of preventive and management strategies.